Review
| Rev Diabet Stud,
2017,
14(1):39-50 |
DOI 10.1900/RDS.2017.14.39 |
The Nexus of Stem Cell-Derived Beta-Cells and Genome Engineering
Sara D. Sackett, Aida Rodriguez, Jon S. Odorico
Division of Transplantation, Department of Surgery, School of Medicine and Public Health, University of Wisconsin-Madison, 1111 Highland Ave, Madison, WI 53711, USA
Address correspondence to: Sara Dutton Sackett, email: sackett@surgery.wisc.edu
Manuscript submitted June 11, 2017; accepted June 11, 2017.
Keywords: diabetes, beta-cell, islet, CRISPR, pluripotent stem cell, GWAS, autoimmunity, allograft rejection, HLA molecules, custom-engineered nuclease
Abstract
Diabetes, type 1 and type 2 (T1D and T2D), are diseases of epidemic proportions, which are complicated and defined by genetics, epigenetics, environment, and lifestyle choices. Current therapies consist of whole pancreas or islet transplantation. However, these approaches require life-time immunosuppression, and are compounded by the paucity of available donors. Pluripotent stem cells have advanced research in the fields of stem cell biology, drug development, disease modeling, and regenerative medicine, and importantly allows for the interrogation of therapeutic interventions. Recent developments in beta-cell differentiation and genomic modifications are now propelling investigations into the mechanisms behind beta-cell failure and autoimmunity, and offer new strategies for reducing the propensity for immunogenicity. This review discusses the derivation of endocrine lineage cells from human pluripotent stem cells for the treatment of diabetes, and how the editing or manipulation of their genomes can transcend many of the remaining challenges of stem cell technologies, leading to superior transplantation and diabetes drug discovery platforms.
Abbreviations: APC antigen-presenting cell; APM antigen-processing machinery; ARX aristaless related homeobox; β2m beta-2-microglobulin deficient; CDKAL1 cyclin-dependent kinase 5 regulatory subunit associated protein 1; ChIP chromotin immunoprecipitation; CNVs copy number variants; CRISPR clustered regularly interspaced short palindromic repeats; CRISPR associated (Cas); CTLA-4 cytotoxic T lymphocyte associated-4; dCas9 catalytically inactive Cas9; DCs dendritic cells; DCregs DC regulatory; ESCs embryonic stem cells; GLIS3 GLI-similar 3; GWAS genome-wide association studies; HDR homology-directed repair; HES1 hairy enhancer of split; HLA human leukocyte antigens; hPSC human pluripotent stem cell; HSV-TK herpes simplex virus - thymidine kinase; iPSCs induced pluripotent stem cells; JNK c-Jun N-terminal kinase; MafA muscloaponeurotic fibrosacrcoma A; MNX1 motor neuron and pancreas homeobox 1; MODY maturity diabetes of the young; MSC mesenchymal stem cell; MT metallothionein; NEUROD1 neurogenic differentiation 1; NEUROG3 neurogenin 3; NHEJ non-homologous end joining; NOD non-obese diabetic; PD-1/PD-L1 programmed cell death; PDX1 pancreatic and duodenal homeobox 1; PTPN22 protein tyrosine phosphatase non-receptor type 22; PTF1A pancreas transcription factor 1A; RFX6 regulatory factor X6; STZ streptozotocin; TALEN transcription activator-like effector nucleases; TCR T cell receptor; TNFAIP3 TNF-induced protein 3; Treg regulatory T cell; T1D type 1 diabetes; T2D type 2 diabetes; ZFN zinc-finger nucleases
1. Introduction
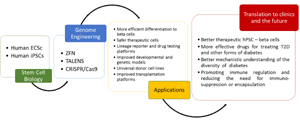
Human pluripotent stem cell (hPSC) therapy has been envisioned for use in regenerative medicine since its conception as a surrogate source of beta-cells [1, 2]. Scientists have considered the potential of PSCs for the treatment of diabetes for nearly two decades, but until recently the inability to achieve a functional and enriched beta-cell mass stymied advancement in the field. Recent progress in deriving functional beta-cells [3, 4] from human PSCs significantly advances the goal of achieving an effective, transplantable beta-cell mass for the many patients with insulin-dependent diabetes closer to reality. Additionally, hPSC-beta-cells provide a unique and valuable platform for drug screening and studying human pancreatic beta-cell biology. Beyond improved functionality, challenges remain in fulfilling the clinical and scientific potential of hPSC-beta-cells, such as those related to the control of heterogeneity, tumorigenicity, and immunogenicity of the final cell product, which could potentially be surmounted through genome engineering. For example, efficient drug screening platforms require reproducible, standardized cell populations and rapid, simple reporter assays. In addition, using cells to study pancreatic beta-cell development and function requires genetic manipulation to alter gene product expression. The ability to perform controlled genetic modifications, such as inserting a gene reporter, replacing a normal gene with a mutated gene, inserting a selectable marker or eliminating HLA antigens, has enormous potential to amplify the power of hPSC-beta-cells for both clinical and scientific applications, but until recently, manipulation depended labor-intensive and inefficient standard transfection and homologous recombination techniques. Recent progress in genome editing [5, 6] renders genetic modification much more efficient and multiplexable, yielding modified cells in as fast as several weeks (Figure 1).
 |
 |
Figure 1. Genome engineering technologies. Intersection of powerful genome engineering technologies and stem cells will likely contribute to better diabetes drug discovery assays and transplantable stem cell-derived beta-cells. Abbreviations: ZFN: zinc-finger nucleases; TALEN: transcription activator-like effector nucleases; CRISPR: clustered regularly interspaced short palindromic repeats; Cas: CRISPR associated; ESCs: embryonic stem cells; iPSCs: induced pluripotent stem cells; T2D: type 2 diabetes. |
|
This article surveys the many opportunities for merging hPSC-beta-cell and genome engineering technologies to capitalize on the full potential of hPSCs and overcome current bottlenecks in clinical and scientific applications (Figure 1). This review will not focus on the pitfalls or technical aspects of genome editing, which can be found in several other excellent reviews [7, 8].
2. Programmable modification through endonucleases
The development and introduction of programmable site-specific and custom-engineered nucleases has propelled and facilitated genome editing to new levels. The first site-specific endonuclease applied successfully in hPSC genome editing was zinc-finger nuclease (ZFN) [9, 10]. However, ZFNs are difficult to engineer, and they are technically challenging to design and construct, causing the production to be expensive. Transcription activator-like effector nucleases (TALEN) and the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) systems are the most widely used customized nuclease technologies, which have been developed recently [7]. The nucleases induce double strand breaks in DNA at loci of interest, thereby triggering the endogenous DNA repair machinery through the error-prone non-homologous end joining (NHEJ) or the precise homology-directed repair (HDR). These endogenous cellular repair mechanisms are harnessed to facilitate the introduction or repair of mutations or insertion of DNA elements. The CRISPR/Cas9 system is a simpler, more efficient, and cost-effective approach, which utilizes sequence-specific single short-guide RNAs to direct the Cas9 endonuclease to virtually anywhere in the genome. Almost any cell type can be targeted, but most commonly stem cells, including human and murine pluripotent stem cells, are modified for in vitro studies and animal model production. These developments have made the process of manipulating the genome much easier and applicable to a wide range of cell types and organisms [11-13].
3. Lineage reporters for purification and drug discovery platforms
A feared complication of the transplantation of PSC-derived immature or mature therapeutic cell populations, including PSC-derived beta-cells is the development of a teratoma or benign tumor containing primitive cell types derived from all three germ layers. Malignant transformation of the transplanted cells is also a possibility, though considered rarely. It is widely thought that the more differentiated a cell population the lower the number of residual undifferentiated PSCs remaining in the transplanted cell population and the lower the teratoma risk, since studies have shown that teratoma formation is related to the residual dose of undifferentiated cells [14, 15]. Interestingly, studies have also documented that a malignant potential may arise because of the selection of aneuploid clones and other mutations that can accumulate during suboptimal growth conditions [16-18]. Antibody-based negative selection methods [14, 15] and chemical ablation methods [16, 19, 20] have been devised to reduce tumorigenicity by removing undifferentiated cells from immature cell populations. However, these methods are cumbersome as they generally rely on multiple steps and potential off target effects, and it is not clear whether these approaches are applicable in vivo for removing cells in a patient. Even if undifferentiated residual teratomatous cells are removed prior to transplantation, the other mechanisms of malignant transformation remain at play [21], and cancer development in the transplanted cell population is still a risk.
Moreover, despite the fact that recent beta-cell and islet-like cluster protocols appear to have a very low "reported rate" of forming cancers or teratomas [3, 4, 22], the risk is likely heightened when considering the aim of a long-term therapeutic effect. Thus, it may be necessary to physically or genetically construct a failsafe means of killing hPSC-derived cells should a benign or malignant tumor arise. Insertion of a suicide gene, such as herpes simplex virus thymidine kinase (HSV-TK), which would make cells vulnerable to ganciclovir, would be a way to insure an ability to kill all hPSC-derived cells, while leaving normal mammalian somatic cells unaffected (which do not express HSV-TK) should cells go rogue in a particular patient. This approach has been applied successfully in experimental systems [23]. Ganciclovir is well tolerated by patients; its safe clinical use has been extensively documented. Another suicide gene approach is the elimination of hPSCs and their progeny by the inducible expression of a Caspase-9 suicide gene that is activated by a specific chemical inducer of dimerization [24, 25]. Although these strategies do not use genome editing to achieve expression, either CRISPR/Cas9 or TALEN-mediated editing can be used to insert these genes under constitutive promoters into the safe harbor AAVS1 locus or into an endogenous locus using a specific promoter. Moreover, the ease of achieving multiple modifications with genome editing facilitates insertion of more than one suicide failsafe mechanism.
Another issue with current protocols of stem cell-derived islet-like clusters is that other unwanted cell types may be present due to inefficient and asynchronous differentiation. A means to enrich populations of interest which has been repeatedly used in the derivation of myriad cell types, including pancreatic endocrine lineage cells, is based on the concept of using beta-cell transcription factor and insulin-based lineage reporters [26-29]. Genome editing makes genetic transformations, which involve homologous recombination-mediated insertion of a transcription factor or endocrine gene promoter linked to a reporter, readily achievable.
To date, single reporters are used primarily in pancreatic and other lineages within the stem cell field, which limits their experimental utility. For example, during the differentiation process of hPSCs to endocrine lineages, one commonly obtains a subpopulation of polyhormonal cells expressing both insulin and glucagon, contaminating more mature monohormonal cells. Thus, a single reporter would select both monohormonal cells and the less desirable polyhormonal cells, and would not be suitably discriminatory. The multiplexability of CRISPR/Cas9 affords the opportunity to insert multiple reporters, which could allow for more specific positive and negative selection strategies. Reporter molecules have been based on fluorescent proteins (e.g. eGFP) or ectopic expression of cell surface proteins that can be recognized by well-characterized antibodies [30] or expression of antibiotic resistance genes. Each has advantages and disadvantages for clinical applications. In the experimental situation, however, these considerations are generally less germane, and so reporter-based enrichment or selection techniques could be used to enhance the study of specific cell types in developmental, drug discovery, and drug toxicity studies, among others.
Most reporters select cells based on the production of a protein, or what is referred to as phenotypic selection. A novel functional cell reporter system was recently devised and illustrated by Burns et al. [31]. This ingenious reporter system linked the luciferase gene to the C-peptide portion of the proinsulin gene. After stably transfecting this construct into a mouse insulinoma cell line, they comprehensively demonstrated the ability to measure functional responses to secretagogues and glucose as well as insulin secretory inhibitors in culture in a highly quantitative manner using a bioluminescence assay on culture supernatant. Such a system, if employed in hPSCs, potentially using genome editing to efficiently transform cells, would provide a nearly ideal high-throughput readout method for measuring functional hPSC-derived beta-cells and insulin secretory responses to new and existing drugs.
4. Genome-editing strategies for immune intervention
A major obstacle in the development of alternative approaches for repair or replacement of organs and tissues is immunogenicity. The therapeutic potential of hPSCs for deriving cells and organs for regenerative medicine has driven intense research over the last 20 years [32, 33]. PSCs broadly comprise either embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) [34, 35], and relative to an intended recipient, PSCs can either be allogeneic (ESCs or iPSCs) or syngeneic (iPSCs). iPSCs have the attribute of being patient-specific, thereby avoiding, potentially, many of the immunogenic properties that allogeneic transplants hold. However, whether autologous iPSC-based therapies would escape immune recognition and destruction by a recipient's immune system is still an unanswered question that needs further investigation in long-term studies [36-38]. As the field progresses, combining iPSC technologies with genomic modification to ensure success in transplantation without the need for immunosuppression brings these types of therapies closer to a clinical reality.
4.1 Genetic editing of HLA types to broaden the application
It is now well established in many laboratories that human pluripotent stem cells (hPSC) can be directed in vitro to differentiate into beta-like insulin-secreting cells. However, until recently, the majority of these insulin-positive cells appeared to be phenotypically and functionally immature (i.e., they have dual hormone expression, low MafA expression, and low insulin secretion in response to glucose) [1, 3, 39-41]. The recent derivation of more mature glucose-responsive beta-cells from hPSCs in vitro [3, 4] brings stem cell-based therapy for diabetes one step closer to a clinical reality, and would rapidly solve the clinical problem of insufficient donor supply for the millions of patients with type 1 diabetes (T1D). Yet, a significant clinical challenge is in front of, namely to overcome allo- and autoimmunity, thereby potentially eliminating the need for chronic immunosuppression, which is associated with significant beta-cell dysfunction and diabetogenicity, among other risks such as increased probability of infection and malignancy. Autologous human iPSCs derived from a T1D patient's blood or skin could provide a cell source that would potentially avoid allograft rejection once transplanted. However, several studies have demonstrated that syngeneic iPSC may indeed be rejected based on the expression of neoantigens and/or epigenetic changes, which could alter the differentiation capacity and/or immunogenicity [42-45]. Thus, the full range of the immune responses to iPSC-derived beta-cells has not been resolved in the context of auto- and alloimmunity.
4.2 Modifying HLA to disrupt immune reaction
Matching human leukocyte antigens (HLA) between a donor and a transplant recipient is desirable, yet due to the large number of polymorphisms associated with the HLA loci, it is difficult to find an exact match [46]. Undifferentiated hESCs express very low levels of HLA class I (HLA-A, -B, and -C in humans), class II (HLA-DR, -DQ, and -DP in humans), and co-stimulatory molecules CD80 and CD86. Because of the observation in older studies that undifferentiated ESCs fail to elicit an immune response in immune competent mice, ESCs were considered immune privileged [47-51]. In contrast to low class I expression on undifferentiated hESCs, HLA class I protein is expressed by most somatic cells [49]. HLA class I molecules expressed on somatic cells engage the T cell receptor (TCR) on CD8+ cytotoxic T effector cells leading to graft damage. Ligation of the T cell TCR to MHC class I in mice was shown to be required for islet allograft destruction, as wild-type allogeneic islets transplanted into beta-2-microglobulin-deficient (β2m) mice were not rejected. This result was due to the absence of cell surface expression of HLA class I (β2m is required for class I cell surface expression) and CD8+ T cells due to failure of proper thymic selection during T cell development [52]. Furthermore, probing the opposite experiment, several groups showed that β2m-/- islets transplanted into wild-type allogeneic mice exhibited prolonged graft survival and minimal islet destruction [53, 54].
However, differentiation to pancreatic lineages results in increased expression of HLA class I, and it is also well known that exposure to an inflammatory environment (in vivo or in vitro) increases class I expression. Thus, the possibility that these influences would promote the upregulation of these molecules rendering them vulnerable to immune-mediated damage such as acute rejection is raised [48, 50, 55-58]. Van der Torren et al. recently investigated the immunogenicity of stem cell-derived beta-cells and their progenitors to adaptive immune responses. They demonstrated that when hESCs are differentiated towards pancreatic endoderm cells, they maintain low levels of HLA class I proteins. This means that these progenitor cells maintain a hypoimmunogenic state when protected from exposure to cytokines, such as IFNγ, i.e., in a non-inflammed environment. However, when these cells are further differentiated towards endocrine cells, HLA antigens were upregulated, thereby making these cells targets for destruction by cytotoxic T lymphocytes and antibody-dependent cellular toxicity. These results underline the importance of protecting transplanted grafts through encapsulation and further suggest that the hypoimmunogenicity found in pancreatic endoderm can be manipulated to induce graft-specific tolerance [51].
4.3 Inducing tolerance through genomic editing
Immune intervention via controlling the expression of HLA class I and II genes through genomic modifications is therefore an interesting approach for providing a graft with the ability to escape immune recognition and destruction. As mentioned above, hESCs exhibit low immunogenicity, but investigating the mechanisms that regulate HLA gene expression is a necessary step in developing strategies to induce tolerance. Suarez-Alvarez et al. presented data on investigating expression levels of classical and non-classical MHC class I and II molecules, as well as antigen-processing machinery (APM) components through bisulfate sequencing and chromotin immunoprecipitation (ChIP) assays for the analysis of posttranslational modifications placed on the histones [50]. They observed that the absence or low level of MHC expression in hESC was due to a lack of APM gene expression. During the differentiation process, these genes were upregulated, which led to an increased presence of MHC class I. They concluded that these processes were regulated by modifications in chromatin remodeling, specifically in H3K4me3 in HLA-B and β2m. This study demonstrated evidence for the role of epigenetic modifications in the regulation and control of MHC class I and II in hPSCs and iPSCs. If the mechanisms can be further understood, then hPSCs and iPSCs could be manipulated to induce tolerance [50].
Genome editing techniques enable the exploration of the hypothesis that removal of all HLA antigen expression from hPSCs could significantly retard or prevent the allorecognition of, and adaptive immune responses to transplanted allogeneic stem cell-derived cells. Using ZFN to modify HLA expression, Torikai et al. sought to modify hematopoietic stem cells (HSC) to broaden and improve transplants in patients with hematologic disorders. By eliminating expression of HLA-A complex, they demonstrated that these cells maintained the ability to engraft in immunocompromised mice. Furthermore, there was an increased chance of finding HLA-matched donors by deleting these regions [59]. These results highlight the possibility for generating applications which avoid immune recognition by nature killer cells.
4.4 Dendritic cells and regulatory dendritic cells
The cells of the innate immune system (including neutrophils, macrophages, dendritic cells, and monocytes) provide us with the first line of defense against pathogens and infections through interaction with the adaptive immune system. These cells do not express antigen-specific receptors. However, activated dendritic cells (DCs) stimulate the adaptive immune system through release of cytokines and by acting as costimulatory antigen-presenting cells (APCs) for T cells. There are several interesting facets to the use of PSCs, such as the recognition that a key factor with these hPSC-derived tissues is that endogenous DCs, normally found in transplanted organs, are absent, which could potentially reduce the occurrence of allosensitization [60]. However, it has been shown that autologous tolerogenic DCs can induce Tregs, Th2, and regulatory B cells to promote tolerance [61, 62]. Therefore, combining hPSC-derived tissue, typically lacking DCs, with toleragenic DCs is a potential immunotherapeutic means to induce donor-specific unresponsiveness and prevent rejection. Recently, Cai et al. generated DC regulatory (DCregs) cells from murine iPSCs, and demonstrated that donor-type iPSC-derived DCregs triggered TGFβ secretion, which caused naïve CD4 T cells to differentiate into donor-specific Tregs instead of T effector cells [63]. It is clear that innate immune cells play a role in the development and propagation of immune tolerance and in autoimmune processes [64].
4.5 CTLA-4
Transplantation biology has studied numerous cell surfaces and secreted molecules in order to find mechanisms to elude immune-mediated rejection and promote long-term engraftment of allogeneic cells. A non-HLA gene also associated with T1D is cytotoxic T lymphocyte-associated 4 (CTLA-4). The costimulatory blockade has emerged as a critical mechanism of antigen-specific T cell unresponsiveness, which is mediated through negative signaling via the T cell receptor CTLA-4. CTLA-4 encodes a costimulatory protein, which is expressed on the surface of activated T cells, and competes with CD28 to bind B7 molecules found on APCs providing so-called "signal 2" completing T cell activation [65]. However, CTLA-4 has a higher affinity for B7 than does CD28, which leads to inhibition of T cell function by blocking CD28-mediated activation via ligation to CD80 and CD86 (aka B7-1 and B7-2, respectively). Therefore, CTLA-4 plays an important role in T-cell-mediated autoimmunity related to autoimmune diseases such as T1D [66].
4.6 PD-1/PD-L1 pathway
Another major receptor-ligand network is the programmed cell death (PD-1/PD-L1) pathway, which is an immune inhibitory pathway that restrains T cell activity when activated [67, 68]. PD-L1 on APCs and tumor cells binds to PD-1 receptor expressed on T cells, thereby inhibiting T cell activity [69, 70]. Recent experimental results by Rong et al. suggest that constitutive dual PD-L1 and CTLA4Ig overexpression in hPSC downregulates immune responses to undifferentiated cells and other differentiated progeny such as hPSC-derived cardiomyocytes and fibroblasts [71]. Moreover, El Khatib et al. overexpressed a PD-L1-CTLA4Ig fusion polyprotein in human islets using adeno-associated viral-mediated gene delivery under control of the insulin promoter, and found that PDL1-CTLA4Ig-expressing islets were protected from rejection. They also found that delivery of the fusion protein gene to NOD mice prevented the development of diabetes [72].
4.7 Zinc-finger protein
A20 is a zinc finger protein that is upregulated by cytokines (IL-1β and TNFα) in beta-cells in an NF-ΚB dependent manner. A20 has been shown to protect beta-cells from cytokine-mediated apoptosis. This anti-apoptotic effect is mediated through inhibition of NF-kB and nitric oxide production [73], and via inhibition of the c-Jun N-terminal kinase (JNK) and augmentation of the Akt survival pathways [74]. The study by Fukaya et al. employed genome-wide association studies, and identified the gene TNF-induced protein 3 (TNFAIP3), encoding for A20, as a susceptibility locus for T1D, providing clinical evidence for the development of T1D [74].
4.8 Benefits of modulating the expression of molecules on hPSC-derived endocrine cells
Using genome editing techniques to produce specific changes to the expression of immunomodulatory molecules expressed on the hPSC-derived endocrine cells would allow researchers to study the effects of specific perturbations, which, in combination with humanized mice, could provide important guideposts to ultimately reducing the immunogenicity of the transplanted cells in humans. The generation of hypoimmunogenic and universally compatible cells lines for the generation of tissues and organs for regenerative medicine will be a critical hurdle to clear in order to attenuate the T cell-mediated rejection of transplanted stem cell-derived tissue grafts.
5. Modification of the genome to study the genetic basis of diabetes and beta-cell dysfunction in disease and development
5.1 Neurogenin 3
In the field of diabetes, the use of genome editing techniques is being used to better understand pancreatic development using in vitro hPSC differentiation models. For example, the role of neurogenin 3 (NEUROG3), a known essential endocrine commitment transcription factor in murine systems implicated in the commitment of pancreatic progenitor to islet endocrine cell fates [75-77], has recently been interrogated using inducible CRISPR-Cas9 technology. Inducible NEUROG3 overexpression in hPSC increased the numbers of beta-cells [78], and NEUROG3-/- cells could not mature into endocrine lineages [79]. RFX6 is another key transcription factor in pancreatic lineage specification, which causes neonatal and childhood-onset diabetes [80-82]. A recent study using CRISPR-Cas9 to knockout RFX6 in hESCs showed delayed pancreatic progenitor formation from stem cells in vitro through PDL1 induction [78].
5.2 GATA4 and GATA6
Haploinsufficiency is recognized as an important factor in human diseases such as diabetes. GATA6 haploinsuffiency is implicated in neonatal and adult-onset diabetes, but it is not well-understand so far how GATA6 affects human pancreatic development. In contrast, mice harboring GATA6 heterozygous mutations have normal pancreatic function [83]. To resolve these differences, Shi et al. have generated isogenic GATA6 and GATA4 mutant hPSC lines using CRISPR-Cas9 technology to investigate the role of this gene in pancreatic differentiation. They compared wild-type to mutant hPSC lines in their ability to differentiate into islet-like cells [84], and found that GATA6 and GATA4 were expressed during human endoderm and pancreas differentiation, while GATA6 was required for efficient formation of definitive endoderm and pancreatic progenitor specification as well as in glucose-responsive beta-cells. Furthermore, GATA4 gene dosage was also implicated in the formation of pancreatic progenitors [85]. In another recent publication by the same group, they used CRISPR-Cas9 and TALEN gene editing on differentiated hPSC to study the role of pancreatic transcription factors like PDX1, RFX6, PTF1A, GLIS3, MNX1, NGN3, HES1, and ARX in T2D. This analysis has helped to elucidate the role of RFX6 in the regulation of pancreatic progenitors, a dosage-sensitive requirement for PDX1 in pancreatic endocrine development, and a potentially different role of NGN3 between humans and mice [78]. These studies illustrate how combining stem cell-derived beta-cells and genome editing provides a powerful model system to better understand the human pancreas development. Further studies analogous to these focusing on additional genes are likely to reveal additional novel information for the field.
5.3 Diabetes and animal models – mesenchymal stem cells and CRISPR/Cas9
T2D, maturity diabetes of the young (MODY), and monogenic forms of diabetes are incompletely understood, genetically and mechanistically. While mouse models have provided key insights, they do not fully capture the spectrum of the diseases in humans, and in many cases, the human phenotype is discordant to that seen in mouse models. The use of genome engineering in hPSCs is revolutionizing human genetics, and permitting scientists to determine whether disease associated genetic mutations are causative or correlative [83].
Human clinical trials to treat T1D are currently in progress involving the infusion of mesenchymal stem cells (MSCs) and MSC-derived insulin-producing cells. This approach is postulated to be effective because of the differentiation capabilities and immunomodulatory properties of MSCs. In fact, MSCs have been shown to be effective as an immunomodulatory therapy, and corrected diabetes in both NOD and streptozotocin (STZ) mouse models [86-88]. Findings in these models suggest that MSCs can migrate to areas of pancreatic injury, and modify the microenvironment, promoting survival and regeneration of the remaining beta-cells, while inhibiting autoimmunity targeting regenerating beta-cells [89]. In addition, it is plausible that insulin-producing cells generated from MSCs could retain some MSC properties, and regulate the immune response. A recent review proposed the use of CRISPR and short guide RNAs to target endogenous activation of pancreatic development transcription factors (PDX1, Neurod1, MafA, etc.) and MSC chemokine receptors in MSC-derived insulin-producing cells. If possible, this strategy would allow MSC-derived insulin-producing cells to maintain some immunomodulatory properties in vitro and in vivo, which could enhance the potency of MSCs in T1D therapies [90].
Another study highlighted a way to activate insulin expression in cells that normally do not express it, using CRISPR/Cas9 to modulate epigenetic marks such as chromatin remodeling, DNA methylation, and histone modifications associated with gene activation, which are relevant to the pathogenesis of diabetes [91, 92]. This group focused on whether CRISPR technology can be applied to all cell types, paying attention to the methylation status of the target promoter, and ability to modulate the chromatin state. The study focused on the endogenous human insulin (INS) gene, which is a silenced gene when the promoter is fully methylated. They utilized an expression plasmid consisting of dCas9 (catalytically inactive Cas9) and the transcriptional activator VP160 to drive expression of endogenous genes [93]. They could activate the endogenous human INS gene and drive expression in diverse cell lines, regardless of the methylation status of the INS promoter [91]. In summary, using genome-editing techniques, it may be possible to target genes implicated in T1D and beta-cell function.
5.4 Genome-wide association studies
Genome-wide association studies (GWAS) have increased our knowledge of loci associated with diabetes. However, in many cases, these are simply associations of which we have a poor mechanistic understanding. Some variants are single nucleotide polymorphisms (SNPs), whereas others are insertions/deletions or copy number variants (CNVs). As a consequence, it is sometimes difficult to find out which gene in a determined locus is causative for the disease [94]. To illustrate the potential of combining GWAS-identified susceptibility genes for T2D information with hESCs, Zeng et al. focused on gaining some insight into the mechanisms underlying the genetic variants and disease phenotype. They focused on 3 genes, identified in GWASs, CDKAL1, KCNQ1, and KCNJ11 to systematically examine the role of these genes in the development of T2D. These mutations did not impair the stepwise in vitro differentiation to insulin-producing cells, but demonstrated defects in insulin secretion, both in vitro and in vivo, generating defective glucose homeostasis rather than insulin resistance [95]. In correlation with these results, Zeggini et al. indicated that their findings further demonstrated the association of CDKAL1 (cyclin-dependent kinase 5 regulatory subunit associated protein 1), as a high-risk locus in T2D [96]. These studies suggest that CDKAL1, a member of the methylthiotransferase family, is an attractive candidate for a therapeutic target in T2D due to the identification of SNPs from GWAS studies.
The FOS/JUN pathway is a key regulator in cell growth, and is highly upregulated in the absence of CDKAL1. T52244, a small molecule inhibitor of the FOS/JUN activator complex AP1, was shown to prevent apoptosis in CDKAL1 null cells. The inhibition of this pathway either through CRISPR-mediated technology or by using T52244, rescued the inability of CDKAL1-deficient cells to correct glycemia in streptozotocin-diabetic mice [97]. A similar study also focused on CDKAL1, and found that inhibition of CDKAL1 in hESCs-derived insulin-producing cells was implicated in the downregulation of metallothionein (MT) gene products, genes that are also linked to diabetes detected by GWAS studies. The overexpression of MT1E in knockout CDKAL1 cells rescued the hypersensitivity to glucolipotoxicity, and improved pancreatic beta-cell function in vitro and in vivo [98].
Another interesting study probed the potential role of protein tyrosine phosphatase non-receptor type 22 (PTPN22) in autoimmune diabetes. PTPN22 is expressed in hematopoietic cells, but through GWAS a minor allelic form of this gene PTPN22R620W was found to be associated with an increased risk of diabetes in humans. The introduction of this gene in the NOD mouse model using CRISPR/Cas9 technology showed increased insulin autoantibodies concomitantly with an earlier onset and higher penetrance of T1D [99]. The knowledge from GWAS in combination with hESC-derived beta-cells is an invaluable tool to define the specific mechanistic role of genes associated with human diabetes. Furthermore, this combination provides a unique resource to determine the function of disease-associated loci as well to elucidate the molecular mechanism controlling pancreatic beta-like cell function and survival. Extending this concept, it is possible that in the future genome edited hPSCs targeting multiple different GWAS-derived diabetes gene associations could make up a comprehensive drug screening platform to identify new candidate drugs for treating diabetes.
6. Conclusions
The field of beta-cell replacement therapies has been an exciting direction to move in as an alternative to insulin replacement therapy and transplantation through cadaveric means. Some of these advances in cell therapies have entered clinical trials [100]. The ability to combine human pluripotent stem cell-based technology with state-of-the-art gene editing technology brings the field to an intersection that is influencing basic and applied biology research by generating better in vitro disease models, chemical screens, and cell-based therapies.
Disclosures: JSO is scientific co-founder and chair of the Scientific Advisory Board for Regenerative Medical Solutions, Inc. The other authors report no conflict of interests.
References
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 2008. 26(4):443-452. [DOD] [CrossRef]
- Nostro MC, Keller G. Generation of beta cells from human pluripotent stem cells: Potential for regenerative medicine. Semin Cell Dev Biol 2012. 23(6):701-710. [DOD] [CrossRef]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O'Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014. 32(11):1121-1133. [DOD] [CrossRef]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell 2014. 159(2):428-439. [DOD] [CrossRef]
- Liu Z, Liang Y, Ang EL, Zhao H. A New Era of Genome Integration-Simply Cut and Paste! ACS Synth Biol 2017. 6(4):601-609. [DOD]
- Carlson-Stevermer J, Goedland M, Steyer B, Movaghar A, Lou M, Kohlenberg L, Prestil R, Saha K. High-Content Analysis of CRISPR-Cas9 Gene-Edited Human Embryonic Stem Cells. Stem Cell Reports 2016. 6(1):109-120. [DOD] [CrossRef]
- Hendriks WT, Jiang X, Daheron L, Cowan CA. TALEN- and CRISPR/Cas9-Mediated Gene Editing in Human Pluripotent Stem Cells Using Lipid-Based Transfection. Curr Protoc Stem Cell Biol 2015. 34:5B. [DOD]
- Wang H, La Russa M, Qi LS. CRISPR/Cas9 in Genome Editing and Beyond. Annu Rev Biochem 2016. 85:227-264. [DOD] [CrossRef]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 2009. 27(9):851-857. [DOD] [CrossRef]
- Zou J, Maeder ML, Mali P, Pruett-Miller SM, Thibodeau-Beganny S, Chou BK, Chen G, Ye Z, Park IH, Daley GQ, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 2009. 5(1):97-110. [DOD] [CrossRef]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 2013. 14(1):49-55. [DOD] [CrossRef]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013. 8(11):2281-2308. [DOD] [CrossRef]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 2010. 11(9):636-646. [DOD] [CrossRef]
- Ben-David U, Nudel N, Benvenisty N. Immunologic and chemical targeting of the tight-junction protein Claudin-6 eliminates tumorigenic human pluripotent stem cells. Nat Commun 2013. 4:1992. [DOD] [CrossRef]
- Kahan B, Magliocca J, Merriam F, Treff N, Budde M, Nelson J, Browning V, Ziehr B, Odorico J. Elimination of tumorigenic stem cells from differentiated progeny and selection of definitive endoderm reveals a Pdx1+ foregut endoderm stem cell lineage. Stem Cell Res 2011. 6(2):143-157. [DOD] [CrossRef]
- Ben-David U, Benvenisty N. Chemical ablation of tumor-initiating human pluripotent stem cells. Nat Protoc 2014. 9(3):729-740. [DOD] [CrossRef]
- Ben-David U. Genomic instability, driver genes and cell selection: Projections from cancer to stem cells. Biochim Biophys Acta 2015. 1849(4):427-435. [DOD] [CrossRef]
- Heslop JA, Hammond TG, Santeramo I, Tort Piella A, Hopp I, Zhou J, Baty R, Graziano EI, Proto Marco B, Caron A, et al. Concise review: workshop review: understanding and assessing the risks of stem cell-based therapies. Stem Cells Transl Med 2015. 4(4):389-400. [DOD] [CrossRef]
- Ben-David U, Biran A, Scaffidi P, Herold-Mende C, Boehringer M, Meshorer E, Benvenisty N. Elimination of undifferentiated cancer cells by pluripotent stem cell inhibitors. J Mol Cell Biol 2014. 6(3):267-269. [DOD] [CrossRef]
- Bedel A, Beliveau F, Lamrissi-Garcia I, Rousseau B, Moranvillier I, Rucheton B, Guyonnet-Duperat V, Cardinaud B, de Verneuil H, Moreau-Gaudry F, Dabernat S. Preventing Pluripotent Cell Teratoma in Regenerative Medicine Applied to Hematology Disorders. Stem Cells Transl Med 2017. 6(2):382-393. [DOD] [CrossRef]
- Lamm N, Ben-David U, Golan-Lev T, Storchova Z, Benvenisty N, Kerem B. Genomic Instability in Human Pluripotent Stem Cells Arises from Replicative Stress and Chromosome Condensation Defects. Cell Stem Cell 2016. 18(2):253-261. [DOD] [CrossRef]
- Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J 2015. 34(13):1759-1772. [DOD] [CrossRef]
- Tieng V, Cherpin O, Gutzwiller E, Zambon AC, Delgado C, Salmon P, Dubois-Dauphin M, Krause KH. Elimination of proliferating cells from CNS grafts using a Ki67 promoter-driven thymidine kinase. Mol Ther Methods Clin Dev 2016. 6:16069. [DOD] [CrossRef]
- Bedel A, Beliveau F, Lamrissi-Garcia I, Rousseau B, Moranvillier I, Rucheton B, Guyonnet-Duperat V, Cardinaud B, de Verneuil H, Moreau-Gaudry F, Dabernat S. Preventing Pluripotent Cell Teratoma in Regenerative Medicine Applied to Hematology Disorders. Stem Cells Transl Med 2016. In press. [DOD]
- Yagyu S, Hoyos V, Del Bufalo F, Brenner MK. An Inducible Caspase-9 Suicide Gene to Improve the Safety of Therapy Using Human Induced Pluripotent Stem Cells. Mol Ther 2015. 23(9):1475-1485. [DOD] [CrossRef]
- Basford CL, Prentice KJ, Hardy AB, Sarangi F, Micallef SJ, Li X, Guo Q, Elefanty AG, Stanley EG, Keller G, et al. The functional and molecular characterisation of human embryonic stem cell-derived insulin-positive cells compared with adult pancreatic beta cells. Diabetologia 2012. 55(2):358-371. [DOD] [CrossRef]
- Basiri M, Behmanesh M, Tahamtani Y, Khalooghi K, Moradmand A, Baharvand H. The Convenience of Single Homology Arm Donor DNA and CRISPR/Cas9-Nickase for Targeted Insertion of Long DNA Fragment. Cell J 2017. 18(4):532-539. [DOD]
- Porciuncula A, Kumar A, Rodriguez S, Atari M, Arana M, Martin F, Soria B, Prosper F, Verfaillie C, Barajas M. Pancreatic differentiation of Pdx1-GFP reporter mouse induced pluripotent stem cells. Differentiation 2016. 92(5):249-256. [DOD] [CrossRef]
- Williams MD, Wong W, Rixon A, Satoor SN, Hardikar AA, Joglekar MV. Pdx1 (GFP/w) mice for isolation, characterization, and differentiation of pancreatic progenitor cells. Methods Mol Biol 2014. 1194:271-288. [DOD]
- Gadue P, Gouon-Evans V, Cheng X, Wandzioch E, Zaret KS, Grompe M, Streeter PR, Keller GM. Generation of monoclonal antibodies specific for cell surface molecules expressed on early mouse endoderm. Stem Cells 2009. 27(9):2103-2113. [DOD] [CrossRef]
- Burns SM, Vetere A, Walpita D, Dancik V, Khodier C, Perez J, Clemons PA, Wagner BK, Altshuler D. High-throughput luminescent reporter of insulin secretion for discovering regulators of pancreatic Beta-cell function. Cell Metab 2015. 21(1):126-137. [DOD] [CrossRef]
- Odorico JS, Kaufman DS, Thomson JA. Multilineage differentiation from human embryonic stem cell lines. Stem Cells 2001. 19(3):193-204. [DOD] [CrossRef]
- Hoffman LM, Carpenter MK. Characterization and culture of human embryonic stem cells. Nat Biotechnol 2005. 23(6):699-708. [DOD] [CrossRef]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science 1998. 282(5391):1145-1147. [DOD] [CrossRef]
- Yamanaka S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 2007. 1(1):39-49. [DOD] [CrossRef]
- Ruiz S, Diep D, Gore A, Panopoulos AD, Montserrat N, Plongthongkum N, Kumar S, Fung HL, Giorgetti A, Bilic J, et al. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proc Natl Acad Sci U S A 2012. 109(40):16196-16201. [DOD] [CrossRef]
- Stripecke R, Carmen Villacres M, Skelton D, Satake N, Halene S, Kohn D. Immune response to green fluorescent protein: implications for gene therapy. Gene Ther 1999. 6(7):1305-1312. [DOD] [CrossRef]
- Sackett SD, Brown ME, Tremmel DM, Ellis T, Burlingham WJ, Odorico JS. Modulation of human allogeneic and syngeneic pluripotent stem cells and immunological implications for transplantation. Transplant Rev (Orlando) 2016. 30(2):61-70. [DOD] [CrossRef]
- D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 2006. 24(11):1392-1401. [DOD] [CrossRef]
- Rezania A, Bruin JE, Riedel MJ, Mojibian M, Asadi A, Xu J, Gauvin R, Narayan K, Karanu F, O'Neil JJ, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012. 61(8):2016-2029. [DOD] [CrossRef]
- Hrvatin S, O'Donnell CW, Deng F, Millman JR, Pagliuca FW, DiIorio P, Rezania A, Gifford DK, Melton DA. Differentiated human stem cells resemble fetal, not adult, beta cells. Proc Natl Acad Sci U S A 2014. 111(8):3038-3043. [DOD] [CrossRef]
- Zhao T, Zhang ZN, Westenskow PD, Todorova D, Hu Z, Lin T, Rong Z, Kim J, He J, Wang M, et al. Humanized Mice Reveal Differential Immunogenicity of Cells Derived from Autologous Induced Pluripotent Stem Cells. Cell Stem Cell 2015. 17(3):353-359. [DOD] [CrossRef]
- Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature 2011. 474(7350):212-215. [DOD] [CrossRef]
- de Almeida PE, Meyer EH, Kooreman NG, Diecke S, Dey D, Sanchez-Freire V, Hu S, Ebert A, Odegaard J, Mordwinkin NM, et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat Commun 2014. 5:3903. [DOD] [CrossRef]
- Guha P, Morgan JW, Mostoslavsky G, Rodrigues NP, Boyd AS. Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell 2013. 12(4):407-412. [DOD] [CrossRef]
- de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 2006. 38(10):1166-1172. [DOD] [CrossRef]
- Wu DC, Boyd AS, Wood KJ. Embryonic stem cells and their differentiated derivatives have a fragile immune privilege but still represent novel targets of immune attack. Stem Cells 2008. 26(8):1939-1950. [DOD] [CrossRef]
- Li L, Baroja ML, Majumdar A, Chadwick K, Rouleau A, Gallacher L, Ferber I, Lebkowski J, Martin T, Madrenas J, Bhatia M. Human embryonic stem cells possess immune-privileged properties. Stem Cells 2004. 22(4):448-456. [DOD] [CrossRef]
- Drukker M, Katz G, Urbach A, Schuldiner M, Markel G, Itskovitz-Eldor J, Reubinoff B, Mandelboim O, Benvenisty N. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci U S A 2002. 99(15):9864-9869. [DOD] [CrossRef]
- Suarez-Alvarez B, Rodriguez RM, Calvanese V, Blanco-Gelaz MA, Suhr ST, Ortega F, Otero J, Cibelli JB, Moore H, Fraga MF, Lopez-Larrea C. Epigenetic mechanisms regulate MHC and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS One 2010. 5(4):e10192. [DOD] [CrossRef]
- van der Torren CR, Zaldumbide A, Duinkerken G, Brand-Schaaf SH, Peakman M, Stange G, Martinson L, Kroon E, Brandon EP, Pipeleers D, Roep BO. Immunogenicity of human embryonic stem cell-derived beta cells. Diabetologia 2017. 60(1):126-133. [DOD] [CrossRef]
- Desai NM, Bassiri H, Kim J, Koller BH, Smithies O, Barker CF, Naji A, Markmann JF. Islet allograft, islet xenograft, and skin allograft survival in CD8+ T lymphocyte-deficient mice. Transplantation 1993. 55(4):718-722. [DOD] [CrossRef]
- Markmann JF, Bassiri H, Desai NM, Odorico JS, Kim JI, Koller BH, Smithies O, Barker CF. Indefinite survival of MHC class I-deficient murine pancreatic islet allografts. Transplantation 1992. 54(6):1085-1089. [DOD] [CrossRef]
- Osorio RW, Ascher NL, Jaenisch R, Freise CE, Roberts JP, Stock PG. Major histocompatibility complex class I deficiency prolongs islet allograft survival. Diabetes 1993. 42(10):1520-1527. [DOD] [CrossRef]
- Liu P, Chen S, Li X, Qin L, Huang K, Wang L, Huang W, Li S, Jia B, Zhong M, et al. Low immunogenicity of neural progenitor cells differentiated from induced pluripotent stem cells derived from less immunogenic somatic cells. PLoS One 2013. 8(7):e69617. [DOD] [CrossRef]
- Wang X, Qin J, Zhao RC, Zenke M. Reduced immunogenicity of induced pluripotent stem cells derived from Sertoli cells. PLoS One 2014. 9(8):e106110. [DOD] [CrossRef]
- Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 2010. 28(8):848-855. [DOD] [CrossRef]
- de Almeida PE, Ransohoff JD, Nahid A, Wu JC. Immunogenicity of pluripotent stem cells and their derivatives. Circ Res 2013. 112(3):549-561. [DOD] [CrossRef]
- Torikai H, Mi T, Gragert L, Maiers M, Najjar A, Ang S, Maiti S, Dai J, Switzer KC, Huls H, et al. Genetic editing of HLA expression in hematopoietic stem cells to broaden their human application. Sci Rep 2016. 6:21757. [DOD] [CrossRef]
- Bolton EM, Bradley JA. Avoiding immunological rejection in regenerative medicine. Regen Med 2015. 10(3):287-304. [DOD] [CrossRef]
- Creusot RJ, Giannoukakis N, Trucco M, Clare-Salzler MJ, Fathman CG. It's time to bring dendritic cell therapy to type 1 diabetes. Diabetes 2014. 63(1):20-30. [DOD] [CrossRef]
- Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011. 34(9):2026-2032. [DOD] [CrossRef]
- Cai S, Hou J, Fujino M, Zhang Q, Ichimaru N, Takahara S, Araki R, Lu L, Chen JM, Zhuang J, et al. iPSC-Derived Regulatory Dendritic Cells Inhibit Allograft Rejection by Generating Alloantigen-Specific Regulatory T Cells. Stem Cell Reports 2017. 8(5):1174-1189. [DOD] [CrossRef]
- Hua Z, Gross AJ, Lamagna C, Ramos-Hernandez N, Scapini P, Ji M, Shao H, Lowell CA, Hou B, DeFranco AL. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol 2014. 192(3):875-885. [DOD] [CrossRef]
- Kavvoura FK, Ioannidis JP. CTLA-4 gene polymorphisms and susceptibility to type 1 diabetes mellitus: a HuGE Review and meta-analysis. Am J Epidemiol 2005. 162(1):3-16. [DOD] [CrossRef]
- Kristiansen OP, Larsen ZM, Pociot F. CTLA-4 in autoimmune diseases - a general susceptibility gene to autoimmunity? Genes Immun 2000. 1(3):170-184. [DOD]
- Schaub M, Stadlbauer TH, Sayegh MH. CTLA4Ig: effects on cellular and humoral immunity and macrophage activation. Exp Nephrol 1997. 5(5):370-374. [DOD]
- Judge TA, Tang A, Turka LA. Immunosuppression through blockade of CD28:B7-mediated costimulatory signals. Immunol Res 1996. 15(1):38-49. [DOD] [CrossRef]
- Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev 2008. 224:166-182. [DOD] [CrossRef]
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol 2009. 10(11):1185-1192. [DOD] [CrossRef]
- Rong Z, Wang M, Hu Z, Stradner M, Zhu S, Kong H, Yi H, Goldrath A, Yang YG, Xu Y, Fu X. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell 2014. 14(1):121-130. [DOD] [CrossRef]
- El Khatib MM, Sakuma T, Tonne JM, Mohamed MS, Holditch SJ, Lu B, Kudva YC, Ikeda Y. beta-Cell-targeted blockage of PD1 and CTLA4 pathways prevents development of autoimmune diabetes and acute allogeneic islets rejection. Gene Ther 2015. 22(5):430-438. [DOD] [CrossRef]
- Grey ST, Arvelo MB, Hasenkamp W, Bach FH, Ferran C. A20 inhibits cytokine-induced apoptosis and nuclear factor kappaB-dependent gene activation in islets. J Exp Med 1999. 190(8):1135-1146. [DOD] [CrossRef]
- Fukaya M, Brorsson CA, Meyerovich K, Catrysse L, Delaroche D, Vanzela EC, Ortis F, Beyaert R, Nielsen LB, Andersen ML, et al. A20 Inhibits beta-Cell Apoptosis by Multiple Mechanisms and Predicts Residual beta-Cell Function in Type 1 Diabetes. Mol Endocrinol 2016. 30(1):48-61. [DOD] [CrossRef]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 2000. 97(4):1607-1611. [DOD] [CrossRef]
- Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008. 132(2):197-207. [DOD] [CrossRef]
- Pinney SE, Jaeckle Santos LJ, Han Y, Stoffers DA, Simmons RA. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia 2011. 54(10):2606-2614. [DOD] [CrossRef]
- Zhu Z, Li QV, Lee K, Rosen BP, Gonzalez F, Soh CL, Huangfu D. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell 2016. 18(6):755-768. [DOD] [CrossRef]
- McGrath PS, Watson CL, Ingram C, Helmrath MA, Wells JM. The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes 2015. 64(7):2497-2505. [DOD] [CrossRef]
- Smith SB, Qu HQ, Taleb N, Kishimoto NY, Scheel DW, Lu Y, Patch AM, Grabs R, Wang J, Lynn FC, et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature 2010. 463(7282):775-780. [DOD] [CrossRef]
- Concepcion JP, Reh CS, Daniels M, Liu X, Paz VP, Ye H, Highland HM, Hanis CL, Greeley SA. Neonatal diabetes, gallbladder agenesis, duodenal atresia, and intestinal malrotation caused by a novel homozygous mutation in RFX6. Pediatr Diabetes 2014. 15(1):67-72. [DOD] [CrossRef]
- Spiegel R, Dobbie A, Hartman C, de Vries L, Ellard S, Shalev SA. Clinical characterization of a newly described neonatal diabetes syndrome caused by RFX6 mutations. Am J Med Genet A 2011. 155A(11):2821-2825. [DOD] [CrossRef]
- Lorberbaum DS, Sussel L. Gotta Have GATA for Human Pancreas Development. Cell Stem Cell 2017. 20(5):577-579. [DOD] [CrossRef]
- Shi ZD, Lee K, Yang D, Amin S, Verma N, Li QV, Zhu Z, Soh CL, Kumar R, Evans T, et al. Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction with GATA4 in Human Pancreatic Development. Cell Stem Cell 2017. 20(5):675-688. [DOD] [CrossRef]
- Shi ZD, Lee K, Yang D, Amin S, Verma N, Li QV, Zhu Z, Soh CL, Kumar R, Evans T, et al. Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction with GATA4 in Human Pancreatic Development. Cell Stem Cell 2017. 20(5):675-688. [DOD] [CrossRef]
- Bassi EJ, Moraes-Vieira PM, Moreira-Sa CS, Almeida DC, Vieira LM, Cunha CS, Hiyane MI, Basso AS, Pacheco-Silva A, Camara NO. Immune regulatory properties of allogeneic adipose-derived mesenchymal stem cells in the treatment of experimental autoimmune diabetes. Diabetes 2012. 61(10):2534-2545. [DOD] [CrossRef]
- Ezquer FE, Ezquer ME, Parrau DB, Carpio D, Yanez AJ, Conget PA. Systemic administration of multipotent mesenchymal stromal cells reverts hyperglycemia and prevents nephropathy in type 1 diabetic mice. Biol Blood Marrow Transplant 2008. 14(6):631-640. [DOD] [CrossRef]
- Madec AM, Mallone R, Afonso G, Abou Mrad E, Mesnier A, Eljaafari A, Thivolet C. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia 2009. 52(7):1391-1399. [DOD] [CrossRef]
- Sohni A, Verfaillie CM. Mesenchymal stem cells migration homing and tracking. Stem Cells Int 2013. 2013:130763. [DOD] [CrossRef]
- Gerace D, Martiniello-Wilks R, Nassif NT, Lal S, Steptoe R, Simpson AM. CRISPR-targeted genome editing of mesenchymal stem cell-derived therapies for type 1 diabetes: a path to clinical success? Stem Cell Res Ther 2017. 8(1):62. [DOD]
- Gimenez CA, Ielpi M, Mutto A, Grosembacher L, Argibay P, Pereyra-Bonnet F. CRISPR-on system for the activation of the endogenous human INS gene. Gene Ther 2016. 23(6):543-547. [DOD] [CrossRef]
- Togliatto G, Dentelli P, Brizzi MF. Skewed Epigenetics: An Alternative Therapeutic Option for Diabetes Complications. J Diabetes Res 2015. 2015:373708. [DOD] [CrossRef]
- Hu J, Lei Y, Wong WK, Liu S, Lee KC, He X, You W, Zhou R, Guo JT, Chen X, et al. Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors. Nucleic Acids Res 2014. 42(7):4375-4390. [DOD] [CrossRef]
- Rutter GA, Pinton P. Mitochondria-associated endoplasmic reticulum membranes in insulin signaling. Diabetes 2014. 63(10):3163-3165. [DOD] [CrossRef]
- Zeng H, Guo M, Zhou T, Tan L, Chong CN, Zhang T, Dong X, Xiang JZ, Yu AS, Yue L, et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell 2016. 19(3):326-340. [DOD] [CrossRef]
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007. 316(5829):1336-1341. [DOD] [CrossRef]
- Rutter GA. Modeling Type 2 Diabetes GWAS Candidate Gene Function in hESCs. Cell Stem Cell 2016. 19(3):281-282. [DOD] [CrossRef]
- Guo M, Zhang T, Dong X, Xiang JZ, Lei M, Evans T, Graumann J, Chen S. Using hESCs to Probe the Interaction of the Diabetes-Associated Genes CDKAL1 and MT1E. Cell Rep 2017. 19(8):1512-1521. [DOD] [CrossRef]
- Lin X, Pelletier S, Gingras S, Rigaud S, Maine CJ, Marquardt K, Dai YD, Sauer K, Rodriguez AR, Martin G, et al. CRISPR-Cas9-Mediated Modification of the NOD Mouse Genome With Ptpn22R619W Mutation Increases Autoimmune Diabetes. Diabetes 2016. 65(8):2134-2138. [DOD] [CrossRef]
- Schulz TC. Concise Review: Manufacturing of Pancreatic Endoderm Cells for Clinical Trials in Type 1 Diabetes. Stem Cells Transl Med 2015. 4(8):927-931. [DOD] [CrossRef]
|


)

