Review
| Rev Diabet Stud,
2017,
14(2-3):269-278 |
DOI 10.1900/RDS.2017.14.269 |
The Role of Iron in Type 1 Diabetes Etiology: A Systematic Review of New Evidence on a Long-Standing Mystery
Karen L. Søgaard1, Christina Ellervik2,3,4,5, Jannet Svensson1,3, Steffen U. Thorsen1
1Copenhagen Diabetes Research Center (CPH-DIRECT), Department of Paediatrics, Herlev Hospital, University of Copenhagen, Herlev Ringvej 75, 2730 Herlev, Denmark
2Department of Production, Research, and Innovation; Region Zealand, Alleen 15, 4180 Sorø, Denmark
3Department of Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Blegdamsvej 3B, 2200 Copenhagen N, Denmark
4Department of Laboratory Medicine, Boston Children`s Hospital, 300 Longwood Avenue, 02115, Boston, MA, USA
5Harvard Medical School, 25 Shattuck St, 02115, Boston, MA, USA
Address correspondence to: Karen L. Søgaard, e-mail: karenlouisesoegaard@gmail.com
Manuscript submitted April 18, 2017; resubmitted July 13, 2017; accepted August 24, 2017.
Keywords: type 1 diabetes, iron overload, iron intake, risk factor, insulin, systematic review
Abstract
BACKGROUND: The incidence of type 1 diabetes (T1D) is rising, which might be due to the influence of environmental factors. Biological and epidemiological evidence has shown that excess iron is associated with beta-cell damage and impaired insulin secretion. AIM: In this review, our aim was to assess the association between iron and the risk of T1D. METHODS: A systematic literature search was performed in PubMed and EMBASE in July 2016. Studies investigating the effect of iron status/intake on the risk of developing T1D later were included, and study quality was evaluated. The results have been summarized in narrative form. RESULTS: From a total of 931 studies screened, we included 4 observational studies evaluating iron intake from drinking water or food during early life and the risk of T1D. The quality of the studies was moderate to high assessed via the nine-star Newcastle Ottawa Scale. One out of the four studies included in this review found estimates of dietary iron intake to be associated with risk of T1D development, whereas three studies found no such relationship for estimates of iron in drinking water. CONCLUSIONS: The limited number of studies included found dietary iron, but not iron in drinking water, to be associated with risk of T1D. Further studies are needed to clarify the association between iron and risk of T1D, especially studies including measurements of body iron status.
Abbreviations: BmP-6 - bone morphogenetic protein 6; BMPR - bone morphogenetic protein signaling receptors type 1 and 2; DMT1 - divalent metal transporter 1; GAD - glutamate decarboxylase; GDM - gestational diabetes mellitus; Gpx4 - glutathione peroxidase 4; HCP-1 - heme carrier protein 1; HFE - high iron (Fe); HH - hereditary hemochromatosis; HJV - hemojuvelin; HLA (DR) - human leukocyte antigens (antigen D-related); HO1 - heme oxygenase 1; IAA - islet auto antigen; IRR - adjusted incidence rate ratio; LIP - labile iron pool; NHH - non-hereditary hemochromatosis; NOS - Newcastle-Ottawa Scale; NTBI - non-transferrin-bound iron; OR - adjusted odds ratio; ROS - reactive oxidative species; SD - standard deviation; SIR - standardized incidence rate; SMAD - mothers against decapentaplegic; T1D - type 1 diabetes; T2D - type 2 diabetes; TF - transferrin; TfR1/TfR2 - transferrin receptor 1 and 2; Zip14 - ZRT/IRT-like protein 14
1. Introduction
Type 1 diabetes (T1D) is a chronic disease that arises from an autoimmune, primarily T-cell-mediated, destruction of insulin-producing beta-cells in the human pancreas [1]. T1D is one of the most common chronic diseases in childhood and adolescence [2], and for reasons that remain unknown, the incidence is increasing, which can be observed especially in Western countries [3]. In contrast, the prevalence of high-risk HLA haplotypes is not increasing. This coincidence suggests the existence of strong environmental forces contributing to the development and increasing incidence of T1D [4, 5].
The autoimmune destruction of beta-cells in T1D happens when islet-infiltrating immune cells release pro-inflammatory cytokines, which is believed to contribute to beta-cell dysfunction and death, in part by triggering intracellular formation of reactive oxidative species (ROS) [6, 7]. Beta-cells are very sensitive to oxidative stress, and the generation of ROS in beta-cells may cause oxidative damage and cell death [6, 8].
Iron is a key trace element in metabolism, and is essential for beta-cell insulin secretion [6, 8], but excessive amounts may be toxic to the beta-cell as iron generates ROS by participating in the Fenton reaction [9]. No actual threshold for iron toxicity has been proposed, but in adult populations, beta-cell dysfunction and diabetes have been observed in relation to serum ferritin level >150 μg/l [10, 11]. The normal range in healthy adults is 20-300 μg/l [12].
We aimed to conduct a systematic review of published studies on the association between elevated concentrations of iron parameters or elevated iron intake, and increased risk of T1D in both pediatric and adult populations.
2. Materials and methods
The PRISMA guidelines were used in this systematic review [13].
2.1 Data sources and searches
The protocol, including the full search string, has been registered with the international prospective register of systematic reviews (PROSPERO) (http://www.crd.york.ac.uk/PROSPERO/) under ID Number 42016042680. We conducted a systematic search of published literature in the PubMed and EMBASE databases on July 4, 2016. Our search combined MeSH terms and keywords related to exposure (iron status markers) and outcome (T1D). The search string was as follows:
“Iron OR irons OR ferric compound OR ferric compounds OR ferrous compound OR ferrous compounds OR ferritin OR ferritins OR transferrin OR transferrins OR transferrin receptor OR transferrin receptors OR iron isotope OR iron isotopes OR iron compounds OR hemosiderin OR hemosiderin OR hemosiderosis OR haemosiderosis OR transferrin saturation
AND
Type 1 diabetes OR diabetes mellitus, type 1 OR autoimmune diabetes OR insulin-dependent diabetes mellitus”
2.2 Study selection and eligibility criteria
The potentially eligible records from the systematic search were imported into Covidence (www.covidence.org), and duplicates were removed. Titles and abstracts were screened by two persons (KLS and SUT) for eligibility. If assessments were inconsistent, the opinion of a third author (JS) was obtained. The following inclusion criteria were pre-specified:
- Human studies (clinical trials, cohort, case-control, or cross-sectional studies)
- Measurements of body iron status (e.g. ferritin, transferrin, transferrin saturation, and iron intake)
- T1D or risk for T1D as an outcome of interest
- T1D duration not longer than 6 months before exposure assessment
- English language articles
We excluded animal studies, non-English language articles, non-original papers (letters, editorials, conference abstracts, reviews, or commentaries) and duplicate publications. Other related outcomes (i.e. type 2 diabetes (T2D), gestational diabetes (GDM)) were excluded as well.
2.3 Data extraction and quality assessment
From each study, we extracted the following information: first author’s name, year of publication, study design, number of participants, exposition to iron, country, and the associations between iron and risk of T1D indicated as adjusted incidence rate ratio (IRR) or adjusted odds ratios (OR).
We evaluated and scored the quality of each study using the nine-star Newcastle-Ottawa Scale (NOS), a tool used for quality assessment of non-randomized studies [14]. Studies were evaluated based on selection, comparability, and outcome, and judged by a maximum of nine stars. Scores above five stars indicate moderate to high study quality. The NOS for cohort and case-control studies was retrieved from: http://www.ohri.ca/programs/clinical_epidemiology/nosgen.pdf.
3. Results
We decided not to conduct a meta-analysis in this review because of the small number of eligible studies, which would have complicated the estimation of between-studies variance, an important factor in statistical analysis. Instead, we restricted our review to descriptive counts of occasions where iron exposure was found to be significantly associated with the risk of T1D. We followed a narrative approach to the review, including the use of key tables and figures and the description and summary of the results in a qualitative manner.
3.1 Literature search results
A flow diagram of the literature search and study selection for body iron status markers and T1D is presented in Figure 1. Of the 931 separate abstracts reviewed independently by two authors, 883 were excluded. Of the 46 full-text articles reviewed thereafter, 42 were excluded. A total of four publications, including one retrospective cohort study and three case-control studies, met the inclusion criteria, and were therefore included.
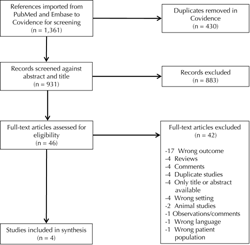 |
 |
Figure 1. Flow diagram on study search. |
|
3.2 Characteristics of the studies included
Table 1 shows the characteristics of the studies included. The number of participants in the studies ranged from 95 to 517. Three studies looked at drinking water components (e.g. iron) as risk factors for T1D, and one study looked at dietary iron intake as a risk factor for T1D. The median bias risk score of the studies included was 7.5 (range: 6-9). The results of the quality assessments are shown in Table 2.
Table
1.
Summary of included studies |
|
 |
|
Legend:
CI - confidence interval, HLA - human leukocyte antigen, IRR - adjusted incidence rate ratio, OR - adjusted odds ratio, SIR - adjusted standardized incidence ratio, T1D - type 1 diabetes. |
|
Table
2.
Study-specific Newcastle-Ottawa quality assessment |
|
 |
|
Legend:
A minimum number of one star (i.e. asterisk) is awarded within category "Selection", and a maximum of two stars within "Comparability". Answers "a" and/or "b" within the sub-categories S1-S4, C1, E1-E3, and O1-O3 meet the requirement for a star. Sub-categories evaluate definitions and selection of cases and controls, representation and selection of cohorts, ascertainment of exposure, and outcome. |
|
3.3 Iron content in drinking water and risk of T1D
A total of three studies focused on the content and amount of trace metals in drinking water at the time of T1D diagnosis [15-17]. This included one retrospective cohort study and two case-control studies of moderate to high assessment quality (6-9). The participants were children with islet autoimmunity [16] or T1D [15-17] diagnosed before the age of 16 and matched controls. The number of participants ranged from 95 to 517 in the cases and from 67 to 323 in the controls [16, 17]. The retrospective cohort study by Zhao et al. had no control population [15]. The studies analyzed the drinking water from the region or municipality in which the children had been resident at the time of diagnosis. In one study, all families received and filled a 200 ml plastic bottle with tap water, which was then analyzed for trace elements including iron [17].
In the two other studies, the data on drinking water compositions were received from the geographically linked water supply [15, 16]. Samuelsson et al. suggested that a high concentration of iron in the water increases the risk of T1D (OR = 1.56, 95% CI = 0.99-2.44), although not significantly [17]. Winkler et al. found that there was no significant difference in the risk of developing islet autoimmunity (IAA) and/or progression of IAA to T1D in children exposed to high vs. low iron concentration in drinking water (OR for development of IAA = 0.8, 95% CI = 0.5-1.2, and OR for progression to T1D = 1.0, 95% CI = 0.4-2.3) [16]. Zhao et al. concluded that iron had no significant relation with the standardized incidence rate (SIR) of T1D [15].
3.4 Dietary iron intake and risk of T1D
One case-control study investigated the effect of iron intake on the development of T1D [18]. The assessment quality of this study was 7. The study found that the OR for developing T1D increased by 2.01 (95% CI = 1.18-3.41) at any increase in standard deviation (SD) of iron intake by one, during the first four months of life, among all participants (128 cases and 67 controls). The OR increased by 2.26 (95% CI = 1.27-4.03) in a restricted sample of T1D cases with control siblings (59 cases and 59 controls).
4. Discussion
This review included a total of four observational studies investigating and reporting results regarding the association between exposure to high iron concentration during early childhood and risk of T1D. The studies found that iron concentrations in drinking water did not have an effect on the risk of developing T1D [15-17], whereas one study found that high dietary iron intake was significantly associated with increased risk of T1D [18].
None of the studies included measured plasma iron status in their study populations, but instead estimated the intake via food or drinking tap water. Iron uptake is tightly regulated, and the absorption of iron intake depends on several factors [19, 20], including the amount of iron, bioavailability of iron from the food source, competing mechanisms at the di-metal luminal transporters, and genetic factors regulating the luminal and basolateral iron transport. However, high absorption of iron may cause an elevated body iron status because excretion mechanisms for iron are limited [19]. As presented in the studies, iron concentration in drinking water was 0.01-0.08 mg/l [15-17], while iron concentration in infant formula was 4.5-12 mg/l [18], demonstrating a substantial difference in iron exposure between the studies. Furthermore, estimation of dietary iron intake may be problematic and possibly confounded. Consequently, measures of iron status would have improved the comparability of the studies. Due to these limitations and the small number of studies on this topic, no conclusions regarding the effect of iron on T1D pathology can be made based on our findings.
The results by Ashraf et al. suggest that elevated iron levels during early life may play a role in the development of T1D. Similarly, a prospective cohort study by Størdal et al. found a significantly higher risk of developing celiac disease, a disease with autoimmune features, in the offspring of mothers treated with an iron supplement during pregnancy [21]. Research confirms the fact that newborns of iron-treated mothers have a higher serum ferritin level than newborns of non-treated mothers [22, 23]. As there is a genetic overlap between celiac disease and T1D [24-26] it seems relevant to investigate whether excess iron, as a result of iron supplementation during pregnancy, could be a risk factor for T1D in the offspring.
Several studies have dealt with iron intake or physiological high iron levels and the risk of other types of diabetes. Two meta-analyses investigating risk factors for T2D have found a strong association between iron and risk of T2D [27, 28]. Also, several studies have found that elevated maternal iron indices and/or dietary iron intake are significantly associated with the risk of GDM [29-33]. Järvelä et al. found that women who develop GDM have an increased risk of both T1D and T2D later in life [34]. This study also found that the majority of patients with GDM who later developed T1D had autoantibodies, reflecting GDM resulting from immune-mediated beta-cell destruction [34]. This demonstrates a potential overlap in the pathogenesis of GDM, T2D, and T1D, and provides another argument for investigating the possible role of iron in T1D pathogenesis.
It is widely documented that extreme iron overload due to conditions such as hereditary hemochromatosis (HH) and non-hereditary hemochromatosis (NHH) is associated with the risk of diabetes [20, 35, 36], and diabetes secondary to these diseases is mimicking both T1D and T2D [37]. Beta-cell destruction in HH and NHH is generally assumed to be a result of iron-mediated ROS production and oxidative stress [6, 8, 20]. However, to the best of our knowledge, no proper classification of diabetes type in HH and NHH, e.g. by examining the prevalence of islet-specific autoantibodies, has been performed.
Experimental animal and human studies support the hypotheses that iron is related to the risk of T1D, and that iron impacts beta-cell function (secretion), insulin sensitivity, and the immune system [20]. Different mechanisms have been proposed:
- Iron contributes to the acceleration of immune initiated beta-cell damage. This theory was presented in a study by Hansen et al., concluding that cytokine-dependent upregulation of divalent metal transporter 1 (DMT1) (Figure 2) and the resulting raise in iron import causes increased ROS-mediated damage in the beta-cell [38]. Furthermore, the study found that knock-out of the DMT1 gene protects against islet inflammation and T1D in a murine model [38].
- Iron accumulation and subsequent tissue damage accelerate or trigger an autoimmune process. This hypothesis is based on a study in hepcidin knock-out mice which found iron accumulation in exocrine pancreas to be accompanied by severe infiltration of macrophages and neutrophils, but no significant difference in lymphocyte content [39]. However, the inflammatory state in the pancreas may potentially initiate an autoimmune response in the parapancreatic lymph nodes by transport of islet-specific autoantigens via professional antigen-presenting cells, thereby triggering T1D.
- Iron overload is widely documented to cause oxidative stress and beta-cell death [6, 20, 38]. However, the mechanism is not fully understood. Ferroptosis is a recently defined non-apoptotic cell death mechanism, which is dependent on iron and ROS formation [40]. Although it has been described in cancer cells almost exclusively, antioxidant enzymes like heme oxygenase 1 (HO1) [41] and glutathione peroxidase 4 (Gpx4) [40, 42] connect ferroptosis to the beta-cell. Therefore, it would be interesting to investigate whether ferroptosis may be an initiator of cell death due to iron overload and oxidative stress in the beta-cell as another pathogenic pathway.
- Iron has a direct effect on autoimmunity. This hypothesis is based on studies showing damage of specific proteins caused by iron-catalyzed ROS, which may lead to beta-cell autoantigens stimulating T cell autoreactivity [43, 44]. This is another mechanism of deregulating self-tolerance in T1D. In this context, autoantibodies from patients with T1D recognize glutamate decarboxylase (GAD) modified by iron-catalyzed oxidation [45]. If this holds true in vivo the autoimmune response may be triggered or accelerated by such epitope modifications.
- Iron influences the number of beta-cells. In a study by Vukicevic et al., the number of Langerhans islets was reduced in mice with knock-out of bone morphogenetic protein-6 (BmP-6) [46], a protein involved in iron homeostasis that is maintained by hepcidin regulation [47] (Figure 2).
- Iron also influences other risk factors of T1D in such way that it affects the progression of T1D development. Iron overload is related to insulin resistance [20, 35], and obesity and insulin resistance have been hypothesized previously to be accelerators of T1D development [48, 49]. It is presumed that the damaged or reduced beta-cells in these conditions are unable to meet the insulin need.
- Finally, beta-cells are found to be an extrahepatic source of hepcidin, with hepcidin stored in and released from the insulin granules [50, 51] (Figure 2). Thus, to some extent, blood glucose may act as a regulator of iron concentration in the blood as increased insulin release will result in hepcidin release and consequently decreased iron release into the circulation. Furthermore, as ferroportin also seems to be located in the membrane of beta-cells [40, 51], the insulin/hepcidin release may result in an autocrine inhibition of iron efflux, thereby increasing the intracellular iron accumulation and ROS formation in beta-cells (Figure 2). We speculate, if this is an additional mechanism behind the association of diabetes with increased food intake (e.g. high glucose) and overweight [52-54], it would support the hypothesis that insulin resistance (and subsequent high blood glucose) accelerates T1D development [48, 49].
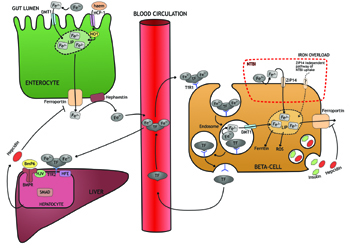 |
|
Figure 2. Several transporters and iron-sensing mechanisms modulate intra- and extracellular iron load. Iron (inorganic and hem-bound) is absorbed from the intestine, and enters the LIP in cytoplasma. Iron is excreted through ferroportin, oxidized by hephestin, and released into the circulation. Iron is transported in the bloodstream bound to transferrin (TF), ensuring controlled delivery of iron to the recipient cell (here beta-cell). On the cell surface, the TF-iron complex binds to transferrin receptor 1 (TfR1) and is internalized into endosomes. Via acidification, iron is released from TF and transported by divalent metal transporter 1 (DMT1) into the labile iron pool (LIP). The unbound TF is released into the bloodstream. In case of iron overload, TF-binding capacity may be exceeded and non-transferrin-bound iron (NTBI) will be present. NTBI is taken up by and transported into the beta-cell in a nonregulated process via ZRT/IRT-like protein 14 (ZIP14) or a ZIP14-independent pathway. Excess iron is stored as ferritin or excreted via ferroportin, but may also cause ROS formation and beta-cell damage (not shown). Hepcidin, secreted by hepatocytes and beta-cells, inhibits iron release by breaking down ferroportin. On the hepatocyte, TF-iron complex binds TfR2 and HFE protein, and by mechanisms not completely understood involving bone morphogenetic protein 6 (BmP6), hemojuvelin (HJV), and mothers against decapentaplegic (SMAD)-mediated intracellular signaling, production of hepcidin is upregulated. In the beta-cell, hepcidin is secreted with insulin and inhibit iron release via ferroportin. Abbreviations: BmP6 - bone morphogenetic protein 6, BMPR - bone morphogenetic protein signaling receptors type 1 + 2, DMT1 - divalent metal transporter 1, HCP-1 - heme carrier protein 1, HO1 - hemeoxygenase 1, LIP - labile iron pool, HJV - hemojuvelin, NTBI - non-transferrin-bound iron, ROS - reactive oxidative species, TF - transferrin, TfR1/TfR2 - transferrin receptor 1 and 2, Zip14 - ZRT/IRT-like protein 14. |
|
It is important to consider that murine models, which were used in the above studies, have been found to be more resistant to the damaging consequences of iron overload [55, 56]. Therefore, more human studies are needed to clarify the effects of excess iron in humans.
In summary, excess iron due to environmental factors or genetic variation is associated with many downstream biochemical and cellular effects on beta-cell function and intracellular ROS formation. Furthermore, it has been proposed that the (high iron) HFE gene has an immunological function independent of iron regulation [57]. The consequences of this function on T1D has not yet been described.
There are several unresolved questions about the relationship of elevated iron indices (or exposure to iron) and risk of T1D:
- Are elevated maternal iron indices (because of iron supplementation during pregnancy, for example) a risk factor for islet autoimmunity or T1D in the offspring?
- Can elevated iron status in the child trigger islet autoimmunity?
- Can elevated iron status in the child influence the progression from islet autoimmunity to T1D during childhood?
- Are there any specific sensitive periods during life where iron might be especially influential?
Owing to the limited quantity of studies included, and the fact that there are no papers dealing with pre-onset measurements of body iron markers and T1D risk in children, further studies are needed, especially pregnancy/offspring cohort studies examining the role of iron intake during pregnancy on mother and offspring, iron levels/status in neonates, and the risk of accelerating and/or developing T1D during childhood. The optimal method for quantifying body iron status is still debated [58]. The "gold-standard" for body iron estimation is liver biopsy, but this invasive procedure is risky and therefore not practicable [59]. Serum ferritin correlates closely with body iron stores in healthy individuals [60, 61], and has a good sensitivity in diagnosing iron deficiency, but it generally fails to rise until iron stores are exceptionally high [62]. In contrast, a lesser degree of iron overload is required to increase transferrin saturation levels [63], which potentially makes transferrin saturation a more suitable choice for estimating physiological iron overload. Of note, the body iron load (intra- and extracellular) is a result of a complex combination of cellular transporters, receptors, importers, and regulative mechanisms (Figure 2) [35, 39, 47, 64], and the interactions between all these components have not yet been fully explored. Therefore, future studies should also include genotyping for polymorphisms in these iron transporters and regulators to identify individuals genetically disposed to harmful effects from moderate degrees of iron overload.
5. Strengths and limitations
One of the strengths of this review is that it is a combined systematic and narrative review. The way it has been conducted is transparent and reproducible because of the systematic literature search and the criterion-based selection of relevant evidence, while the data analysis is conducted in a narrative manner, allowing a more comprehensive approach to study populations and outcomes. Because of the narrow scope of evidence in this field, we found this to be the most appropriate way of presenting this review. Furthermore, we also performed a quality assessment for each study.
The present systematic review is limited as the studies in this field are rare, and most of them are on a relatively small scale. Furthermore, the included studies rely on estimates of iron intake from diet and drinking water only, which may be problematic because of confounding factors. Based on the limited number of studies, a meta-analysis did not seem feasible and the retrospective designs of the studies preclude causal conclusions. However, this emphasizes the need for more research in this field, preferably by performing large prospective cohort studies, e.g. with children at high genetic risk of developing T1D.
6. Conclusions
Iron is an essential trace element, but at moderate to high levels it may be associated with T1D risk. One of four studies included in this review found estimates of dietary iron intake to be associated with the risk of T1D development, whereas three studies found no such relationship for estimates of iron in drinking water.
More studies are needed to clarify the association between iron overload and T1D in humans. Such studies should include sensitive iron status markers and if possible iron-pathway genotyping for in-depth understanding of complex iron-genotype interactions, which could identify specific at-risk groups.
Disclosures: The authors reported no conflict of interests.
References
- Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med 2012. 2(11): a007641. [DOD] [CrossRef]
- Karvonen M, Viik-Kajander M, Moltchanova E, Libman I, LaPorte R, Tuomilehto J. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care 2000. 23(10):1516-1526. [DOD] [CrossRef]
- Gale EA. The rise of childhood type 1 diabetes in the 20th century. Diabetes 2002. 51(12):3353-3361. [DOD] [CrossRef]
- Stankov K, Benc D, Draskovic D. Genetic and epigenetic factors in etiology of diabetes mellitus type 1. Pediatrics 2013.132(6):1112-1122. [DOD] [CrossRef]
- Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR, Gill GV, Gale EA. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet 2004. 364(9446):1699-1700. [DOD] [CrossRef]
- Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 2008. 36(3):343-347. [DOD] [CrossRef]
- Donath MY, Storling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T. Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev 2008. 29(3):334-350. [DOD] [CrossRef]
- Hansen JB, Moen IW, Mandrup-Poulsen T. Iron: the hard player in diabetes pathophysiology. Acta Physiol 2014. 210(4):717-732. [DOD] [CrossRef]
- Sampaio AFS, Silva M, Dornas WC, Costa DC, Silva ME, Dos Santos RC, de Lima WG, Pedrosa ML. Iron toxicity mediated by oxidative stress enhances tissue damage in an animal model of diabetes. Biometals 2014. 27(2):349-361. [DOD] [CrossRef]
- Tuomainen TP, Nyyssönen K, Salonen R, Tervahauta A, Korpela H, Lakka T, Kaplan GA, Solonen JT. Body iron stores are associated with serum insulin and blood glucose concentrations. Population study in 1,013 eastern Finnish men. Diabetes Care 1997. 20(3):426-428. [DOD] [CrossRef]
- Ford ES, Cogswell ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care 1999. 22(12):1978-1983. [DOD] [CrossRef]
- Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk VR, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005. 352(17):1769-1778. [DOD] [CrossRef]
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Int J Surg 2010. 8(5):336-341. [DOD] [CrossRef]
- Newcastle-Ottawa Scale Assessment of Cohort Studies. Pain Management Interventions for Hip Fracture. NCBI Bookshelf Internet. Accessed: Sep 28, 2016. Available from: https://www.ncbi.nlm.nih.gov/books/NBK56664/. [DOD]
- Zhao HX, Mold MD, Stenhouse EA, Bird SC, Wright DE, Demaine AG, Millward BA. Drinking water composition and childhood-onset Type 1 diabetes mellitus in Devon and Cornwall, England. Diabet Med 2001. 18(9):709-717. [DOD] [CrossRef]
- Winkler C, Mollenhauer U, Hummel S, Bonifacio E, Ziegler AG. Exposure to environmental factors in drinking water: risk of islet autoimmunity and type 1 diabetes - the BABYDIAB study. Horm Metab Res 2008. 40(8):566-571. [DOD] [CrossRef]
- Samuelsson U, Oikarinen S, Hyöty H, Ludvigsson J. Low zinc in drinking water is associated with the risk of type 1 diabetes in children. Pediatr Diabetes 2011. 12(3):156-164. [DOD] [CrossRef]
- Ashraf AP, Eason NB, Kabagambe EK, Haritha J, Meleth S, McCormick KL. Dietary iron intake in the first 4 months of infancy and the development of type 1 diabetes: a pilot study. Diabetol Metab Syndr 2010. 2:58. [DOD] [CrossRef]
- Hurrell R, Egli I. Iron bioavailability and dietary reference values. Am J Clin Nutr 2010. 91(5):1461-1467. [DOD] [CrossRef]
- Backe MB, Moen IW, Ellervik C, Hansen JB, Mandrup-Poulsen T. Iron Regulation of Pancreatic Beta-Cell Functions and Oxidative Stress. Annu Rev Nutr 2016. 36:241-273. [DOD] [CrossRef]
- Stordal K, Haugen M, Brantsæter AL, Lundin KE, Stene LC. Association between maternal iron supplementation during pregnancy and risk of celiac disease in children. Clin Gastroenterol Hepatol 2014. 12(4):624-632. [DOD] [CrossRef]
- Milman N, Agger AO, Nielsen OJ. Iron supplementation during pregnancy. Effect on iron status markers, serum erythropoietin and human placental lactogen. A placebo controlled study in 207 Danish women. Dan Med Bull 1991. 38(6):471-476. [DOD]
- Preziosi P, Prual A, Galan P, Daouda H, Boureima H, Hercberg S. Effect of iron supplementation on the iron status of pregnant women: consequences for newborns. Am J Clin Nutr 1997. 66(5):1178-1182. [DOD]
- Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C, Abecasis GR, Barrett JC, Behrens T, Cho J, et al. Pervasive sharing of genetic effects in autoimmune disease. Plos Genet 2011. 7(8): e1002254. [DOD] [CrossRef]
- Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, Howson JM, Stevens H, McManus R, Wijmenga C, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 2008. 359(26):2767-2777. [DOD] [CrossRef]
- Plenge RM. Shared genetic risk factors for type 1 diabetes and celiac disease. N Engl J Med 2008. 359(26):2837-2838. [DOD] [CrossRef]
- Zhao Z, Li S, Liu G, Yan F, Ma X, Huang Z, Tian H. Body iron stores and heme-iron intake in relation to risk of type 2 diabetes: a systematic review and meta-analysis. Plos One 2012. 7(7):e41641. [DOD] [CrossRef]
- Orban E, Schwab S, Thorand B, Huth C. Association of iron indices and type 2 diabetes: a meta-analysis of observational studies. Diabetes Metab Res Rev 2014. 30(5):372-394. [DOD] [CrossRef]
- Bowers K, Yeung E, Williams MA, Qi L, Tobias DK, Hu FB, Zhang C. A prospective study of prepregnancy dietary iron intake and risk for gestational diabetes mellitus. Diabetes Care 2011. 34(7):1557-1563. [DOD] [CrossRef]
- Chen X, Scholl TO, Stein TP. Association of elevated serum ferritin levels and the risk of gestational diabetes mellitus in pregnant women: The Camden study. Diabetes Care 2006. 29(5):1077-1082. [DOD] [CrossRef]
- Khambalia AZ, Aimone A, Nagubandi P, Roberts CL, McElduff A, Morris JM, Powell KL, Tasevski V, Nassar N. High maternal iron status, dietary iron intake and iron supplement use in pregnancy and risk of gestational diabetes mellitus: a prospective study and systematic review. Diabet Med 2016. 33(9):1211-1221. [DOD] [CrossRef]
- Bowers KA, Olsen SF, Bao W, Halldorsson TI, Strøm M, Zhang C. Plasma concentrations of ferritin in early pregnancy are associated with risk of gestational diabetes mellitus in women in the danish national birth cohort. J Nutr 2016. 146(9):1756-1761. [DOD] [CrossRef]
- Fu S, Li F, Zhou J, Liu Z. The relationship between body iron status, iron intake and gestational diabetes: a systematic review and meta-analysis. Medicine (Baltimore) 2016. 95(2):e2383. [DOD] [CrossRef]
- Järvelä IY, Juutinen J, Koskela P, Hartikainen AL, Kulmala P, Knip M, Tapanainen JS. Gestational diabetes identifies women at risk for permanent type 1 and type 2 diabetes in fertile age: predictive role of autoantibodies. Diabetes Care 2006. 29(3):607-612. [DOD] [CrossRef]
- Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab 2013. 17(3):329-341. [DOD] [CrossRef]
- Swaminathan S, Fonseca VA, Alam MG, Shah SV. The role of iron in diabetes and its complications. Diabetes Care 2007. 30(7):1926-1933. [DOD] [CrossRef]
- Cooksey RC, Jouihan HA, Ajioka RS, Hazel MW, Jones DL, Kushner JP, McClain DA. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology 2004. 145(11):5305-5312. [DOD] [CrossRef]
- Hansen JB, Tonnesen MF, Madsen AN, Hagedorn PH, Friberg J, Grunnet LG, Heller RS, Nielsen AO, Storling J, Baeyens L, et al. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic beta cell fate in response to cytokines. Cell Metab 2012. 16(4):449-461. [DOD] [CrossRef]
- Lunova M, Schwarz P, Nuraldeen R, Levada K, Kuscuoglu D, Stützle M, Vujic Spasic M, Haybaeck J, Ruchala P, Jirsa M, et al. Hepcidin knockout mice spontaneously develop chronic pancreatitis owing to cytoplasmic iron overload in acinar cells. J Pathol 2017. 241(1):104-114. [DOD] [CrossRef]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ 2016. 23(3):369-379. [DOD] [CrossRef]
- Jonas JC, Guiot Y, Rahier J, Henquin JC. Haeme-oxygenase 1 expression in rat pancreatic beta cells is stimulated by supraphysiological glucose concentrations and by cyclic AMP. Diabetologia 2003. 46(9):1234-1244. [DOD] [CrossRef]
- Tanaka Y, Tran PO, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A 2002. 99(19):12363-12368. [DOD] [CrossRef]
- Casciola-Rosen L, Wigley F, Rosen A. Scleroderma autoantigens are uniquely fragmented by metal-catalyzed oxidation reactions: implications for pathogenesis. J Exp Med 1997. 185(1):71-79. [DOD] [CrossRef]
- Nerup J, Mandrup-Poulsen T, Helqvist S, Andersen HU, Pociot F, Reimers JI, Cuartero BG, Karlsen AE, Bjerre U, Lorenzen T. On the pathogenesis of IDDM. Diabetologia 1994. 37(Suppl)2:82-89. [DOD] [CrossRef]
- Trigwell SM, Radford PM, Page SR, Loweth AC, James RF, Morgan NG, Todd I. Islet glutamic acid decarboxylase modified by reactive oxygen species is recognized by antibodies from patients with type 1 diabetes mellitus. Clin Exp Immunol 2001. 126(2):242-249. [DOD] [CrossRef]
- Vukicevic S, Grgurevic L. BMP-6 and mesenchymal stem cell differentiation. Cytokine Growth Factor Rev 2009. 20(5-6):441-448. [DOD] [CrossRef]
- Parrow NL, Fleming RE. Bone Morphogenetic Proteins as Regulators of Iron Metabolism. Annu Rev Nutr 2014. 34(1):77-94. [DOD] [CrossRef]
- Wilkin TJ. Diabetes: 1 and 2, or one and the same? Progress with the accelerator hypothesis. Pediatr Diabetes 2008. 9(3):23-32. [DOD] [CrossRef]
- Wilkin TJ. The accelerator hypothesis: weight gain as the missing link between Type I and Type II diabetes. Diabetologia 2001. 44(7):914-922. [DOD] [CrossRef]
- Aigner E, Felder TK, Oberkofler H, Hahne P, Auer S, Soyal S, Stadlmayr A, Schwenoha K, Pirich C, Hengster P, Datz C, Patsch W. Glucose acts as a regulator of serum iron by increasing serum hepcidin concentrations. J Nutr Biochem 2013. 24(1):112-117. [DOD] [CrossRef]
- Kulaksiz H, Fein E, Redecker P, Stremmel W, Adler G, Cetin Y. Pancreatic beta-cells express hepcidin, an iron-uptake regulatory peptide. J Endocrinol 2008. 197(2):241-249. [DOD] [CrossRef]
- van Dam RM, Rimm EB, Willett WC, Stampfer MJ, Hu FB. Dietary patterns and risk for type 2 diabetes mellitus in U.S. men. Ann Intern Med 2002. 136(3):201-209. [DOD] [CrossRef]
- Halton TL, Willett WC, Liu S, Manson JE, Stampfer MJ, Hu FB. Potato and french fry consumption and risk of type 2 diabetes in women. Am J Clin Nutr 2006. 83(2):284-290. [DOD]
- Polsky S, Ellis SL. Obesity, insulin resistance, and type 1 diabetes mellitus. Curr Opin Endocrinol Diabetes Obes 2015. 22(4):277-282. [DOD] [CrossRef]
- Pelot D, Zhou XJ, Carpenter P, Vaziri ND. Effects of experimental hemosiderosis on pancreatic tissue iron content and structure. Dig Dis Sci 1998. 43(11):2411-2414. [DOD] [CrossRef]
- Altamura S, Kessler R, Gröne HJ, Gretz N, Hentze MW, Galy B, Muckenthaler M. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab 2014. 20(2):359-367. [DOD] [CrossRef]
- Reuben A, Chung JW, Lapointe R, Santos MM. The hemochromatosis protein HFE 20 years later: An emerging role in antigen presentation and in the immune system. Immun Inflamm Dis 2017. In press. [DOD]
- Wish JB. Assessing iron status: beyond serum ferritin and transferrin saturation. Clin J Am Soc Nephrol 2006. 1(Suppl 1):S4-S8. [DOD] [CrossRef]
- Cook JD, Finch CA, Smith NJ. Evaluation of the iron status of a population. Blood 1976. 48(3):449-455. [DOD]
- Cook JD, Lipschitz DA, Miles LE, Finch CA. Serum ferritin as a measure of iron stores in normal subjects. Am J Clin Nutr 1974. 27(7):681-687. [DOD]
- Walters GO, Miller FM, Worwood M. Serum ferritin concentration and iron stores in normal subjects. J Clin Pathol 1973. 26(10):770-772. [DOD] [CrossRef]
- Elsayed ME, Sharif MU, Stack AG. Transferrin Saturation: A Body Iron Biomarker. Adv Clin Chem 2016. 75:71-97. [DOD]
- Edwards CQ. Screening for haemochromatosis. J Med Screen 1996. 3(4):170. [DOD] [CrossRef]
- Coffey R, Knutson MD. The plasma membrane metal-ion transporter ZIP14 contributes to 2 non-transferrin-bound iron uptake by human beta cells. Am J Physiol Cell Physiol 2016. In press. [DOD]
|


)
)
)
)

