Diabetic Perspectives
| Rev Diabet Stud,
2005,
2(2):97-109 |
DOI 10.1900/RDS.2005.2.97 |
The Role of HLA Class I Gene Variation in Autoimmune Diabetes
Charles Sia1,  2 2
1Department of Immunology, United Biomedical Inc., 25 Davids Drive, Hauppage, New York 11788, USA.
2Department of Economics, University of Dortmund, 44227 Dortmund, Germany.
Address correspondence to: Charles Sia, e-mail: csia@unitedbiomedical.com
Keywords: type 1 diabetes, HLA class I, cross-study analysis, serotype association
Abstract
The use of DNA-based genetic typing has enabled the identification of type 1 diabetes mellitus (T1DM) susceptible and protective major histocompatibility complex (MHC) class II alleles and haplotypes. The application of this approach has also progressed to locate MHC class I alleles that contribute to the clinicopathology of T1DM. Recent studies have shown a widespread involvement of genes from the MHC class I gene region in the clinicopathology of T1DM. These genes are shown to be involved in contributing to progression from the preclinical stage of the disease, which is characterized by the occurrence of islet-specific antibodies, to clinical disease and also to the occurrence of autoimmunity. They can either contribute directly to disease development or indirectly in concert with other susceptible MHC class II alleles or haplotypes via linkage disequilibrium. Class I alleles may also be negatively associated with T1DM. These findings are useful for the development of future strategies in designing tolerogenic approaches for the prevention or even reversal of T1DM. In this article, the latest evidence for the different kinds of participation of HLA class I genes in the etiology of T1DM is reviewed. A meta-analysis which included existing association studies was also carried out in order to re-assess the relevance of class I genes in diabetes development. The analysis of an enlarged heterogeneous sample confirmed the involvement of previously detected serotypes in the etiology of T1DM, such as A24, B8 and B18, and revealed hitherto unknown associations with B60 and B62. The analysis points out that much of the conflicting results of previous association studies originate from inadequate sample sizes and accentuate the value of future investigations of larger samples for identifying linkage in multigenic diseases.
Introduction
Type 1 diabetes mellitus (T1DM) is a multifactorial and multigenic autoimmune organ disease charac-terized by progressive T cell-mediated destruction of the pancreatic β-cells. The major determinants of this disease are genes of the human leucocyte antigen (HLA) region, which are highly polymorphic. In a multiplicative model, they account for between 20 and 53% of T1DM susceptibility markers [1]. Gene products of HLA class I genes function as antigen presenting molecules for CD8+ cytotoxic T lymphocytes (CTL) and determine the antigen specificity of the CTL-mediated immune response against pathogens and self-antigens. T cell reactions with islet-specific self-antigens may lead to clonal proliferation of autoreactive CTLs directed against pancreatic β-cells provoking destruction of these organ cells.
Initial genetic analyses clearly rejected a dominant major locus. In contrast, a series of other genes, such as the coding sequences for TAP (transporters associated with antigen processing), LMP (large multifunctional proteases) and TNF-α, the MIC-A (MHC class I chain-related A) gene, as well as identified (the insulin gene on chromosome 11p15) and largely unidentified genes on other chromosomes (e.g. 6q and 11q) have been linked to the disease corroborating its polygenic nature [1-5]. This has invoked an additive model, from which it was possible to predict that a maximum of only 0.15% of the general population is at 100% risk of T1DM, about 5% is at intermediate risk, while the remaining population has a risk of almost 0 [6]. The onset of the disease is most common in the young, but is also known to occur at any age, which reflects the heterogeneity of the disease. Early age of onset (≤ 10 yr) is genetically associated with highly susceptible HLA class II alleles from DQA1, DQB1 and DR3 subgroups e.g. the DQA1*0301/DQA1*05011 genotype and the DQA1*0302 allele in a Chinese [7, 8], the DQB1*02/*0302 genotype in a Finnish [9] and the DR3 serotype and A30-B18-DR3 haplotype in a Caucasian population [10]) as well as A*2402 [11]. In contrast, later age of onset (≥ 15 yr) carries haplotypes, which also include protective alleles [9, 12-14]. Generally, young age at onset is associated with rapid progression of disease [15], and very early onset (between the ages of 1 and 2 yr) seems to have a greater genetic and smaller environmental contribution to the initiation of the autoimmune process than at later ages of onset [16].
Clinical appearance is preceded by a preclinical period that may vary from weeks to several years and which involves the occurrence of autoantibodies specific against insulin and other target antigens, such as glutamic acid decarboxylase (GAD) and the tyrosine phosphatase-like islet associated antigen (IA-2), detected in the serum of individuals at risk of developing T1DM. The occurrence of these autoantibodies is considered as evidence for the concomitance of islet-specific autoimmunity and they serve as surrogate markers for T1DM development. In a clinical study including 758 children with newly diagnosed diabetes, 90.7% were positive for two of the four tested antibodies and only 2.1% negative for all four [17]. The induction of antibodies against these self antigens is a T cell-dependent response. In preclinical auto-immune responses antigen presenting cells (APCs) are potentially enabled to present pancreatic self-antigens via MHC class II molecules to CD4+ T cells. These class II/peptide complexes stimulate them by express-ing gene products encoded by disease susceptible HLA class II genes [18]. Progressive damage of β-cells is largely attributed to the cytotoxic activity of diabetogenic CD8+ T cells that infiltrate the pancreatic islets in the latter stages of the cell-mediated immune response [18].
Genetic linkage analyses have also revealed that HLA class I (A, B and C) alleles contribute to T1DM etiology. HLA-A, B and C genes are attributed to conferring risk as well as development of clinical disease with or without the existence of risk DR and DQ genes [19]. It is hypothesized and has recently been verified in NOD mouse models and human genetic case control studies that class I gene products present particular peptides of autoantigen origin to trigger the generation of diabetogenic CD8+ T cells which mediate the progression to T1DM from its preclinical phase to clinical relevance [18, 20, 21]. Although CD8+ T cells seem to participate substantially in the pathogenesis of T1DM, the pathogenic role of certain MHC class I alleles has largely been disregarded [22]. In this article, we therefore aim to discuss the recent findings on classical HLA class I genes and the encoded antigen presentation molecules to re-assess their role in the etiology of T1DM.
Functional impact of HLA class I genes and molecules on the retention of a balanced immunity and their role in T1DM pathogenesis
The primary function of HLA class I molecules is to present peptide fragments derived from cleavage of native antigens in the cytosol. Thus, an explanation for the positive association between T1DM and the gene products encoded by susceptible disease conferring HLA class I genes is their inefficiency to present antigenic peptides required to mediate negative selection for the deletion of autoreactive T cells in the thymus and the periphery. This can result from the architecture of their peptide binding grooves which are in turn determined by their primary sequences. In this regard, the disease susceptible class I molecule A*2402 is related to its non-susceptible A24 counterparts. Their conformational differences may create variability in the peptide binding properties for presentation to CD8+ T cells resulting in the induction of tolerance or self-reactivity [23]. Diversity in the CTL repertoire induced by variations in MHC genes is a critical trait of the immune system to promote powerful anti-pathogenic defense, but it also hazards the consequence of capsizing into an over-aggressive and self-destructing pitfall. Thus T cell selection is a complex system of antagonizing mechanisms equilibrating immune defenses against self- and foreign antigens. In this regard, for instance A24-restricted T cells may be involved in β-cell destruction supported by mechanisms acting in the selection of specific T cell clones. Such mechanisms are benefited by conformational alterations in the antigen-binding pockets due to amino acid substitutions in MHC class I molecules preventing negative selection of autoreactive T cells [23]. The observation that HLA-A24 is associated with rapid progression of T1DM from its preclinical (antibody positive) to its clinical stage characterized by the almost complete destruction of the pancreatic β-cells [24, 25] supports this notion.
The functional mechanisms that may lead to disturbances in the selection of T cells and that direct them to become diabetogenic are still not fully understood. Our presumption that the participation of MHC class I gene variation in the pathogenesis of T1DM has emerged from the observation that enhanced levels of MHC class I heavy-chain RNA are present in pancreatic islets before overt inflammation and the onset of T1DM in the spontaneously diabetic BB rat, but there are no detectable levels of class II antigens [26]. The question of the nature of class I molecules that provide the epitopes restricted to islet-specific autoantigens is of major interest in the understanding of pathways responsible for the activation of β-cell specific autoreactive T cells. Although this question is largely unanswered, it was recently shown that CD8+ CTLs can recognize a peptide from a leader sequence of preproIAPP in A*0201-positive T1DM patients of recent onset [27]. The use of HLA assembly assay has led to the identification of HLA-A*0201-binding peptides that activate GAD-specific CD8+ CTLs in individuals with preclinical T1DM [28]. These CTLs were able to lyse A*0201-positive target cells presenting GAD65 peptide fragments, as pancreatic β-cells do, indicating that class-I-restricted recognition is a critical mechanism even in early stages of the diabetogenic process [28]. A*0201 was found to be positively associated with disease (p = 0.029) in the enlarged sample analyzed within the scope of the meta-study, as shown in the latter section of this article.
Results obtained from genetic studies conducted in the fashion of patients vs. controls have demonstrated that HLA class II genes are clearly associated with the occurrence of high levels of autoantibodies against antigens, such as the 65 kDa isoform of GAD, GAD65 [29], and that these genes contribute decisively to the onset of autoimmunity [7, 9, 13]. The role of class I genes in the early stages of the disease is disputed. In the context of GAD recognition, another indication for the participation of these genes is suggested by the observation that peripheral blood mononuclear cells (PBMC) may respond to GAD peptides with a sequence similar to a protein of Coxsackie B virus. This responsiveness of PBMCs to GAD is certainly not restricted to persons at risk of developing T1DM, but PBMCs of these persons respond significantly more frequently to a sequence encompassing the amino acid residues 247-279 of GAD than healthy persons [30]. Since amino acids 247-279 are encoded by a region that has significant sequence similarity to the P2-C protein of Coxsackie B virus, the clinicopathology of T1DM has long been suspected to be due to molecular mimicry, which in turn substantiates the role of MHC class I genes in preclinical stages of T1DM.
Interestingly, HLA-A2 subgroup molecules were earlier demonstrated to feature high cross-binding propensities to different peptides due to their structural conformity with various residue motifs [31]. Cross-reactivity is a critical phenomenon in cellular immunity where specific MHC epitopes become capable of loading an array of antigenic fragments for the selection and education of T cells for defense against pathogens. In situations where T cells are generated by cross-reactivity of this kind, they become capable of recognizing and reacting even with antigenic peptides other than the original ones that have been used to select these T cells [32]. Indeed peptide cross-binding phenomena were observed for gene products of several MHC class I alleles, such as those of the A2 subgroup and B*2705 [31, 33]. Molecular mimicry and T cell cross-reactivity to β-cell autoantigens and environmental agents with sequence similarities are thus proposed mechanisms that may influence the pathogenesis of type 1 diabetes [34, 35], but has still not been proven to be causal and the mechanism is criticized because T cells could not be shown to cross-react with GAD in vitro [36].
Today's leading concept is that distinct combinations of MHC alleles interact synergistically to induce the disease when further environmental and genetic factors are present [37]. Genetic predisposing alone does not explain the etiology of T1DM as only 40% of monozygotic twins develop the disease concordantly [38]. Thus, the existence of predisposing factors in terms of specific allele variations more or less randomly results in the generation of autoreactive β-cell-specific T cells, namely if other factors, in particular viruses, appear accessorily to induce disturbances of the immunologic equilibrium. In this regard, ex post analyses of diabetic patients, where several factors have already acted in concert to advance the outbreak of the disease, have limited meaning when identifying certain predisposing alleles. It is necessary to also include those persons who are only antibody positive before the clinical manifestation of the disease to identify specific outbreak markers. There is only one study that has tracked the clinical history of antibody positive persons in the context of HLA class I gene susceptibility [18] and the results are reported and re-examined in this article.
Positive association of HLA class I serotypes and alleles with T1DM
Approximately 25 years ago, HLA-B8, B15 and B18 were first found to exist at higher frequencies in diabetic patients of Caucasian origin [39, 40]. Further investigation of these early studies has shown that the B8 serotype association occurs in Caucasians more widely than the B15 and B18 type (Table 1) [18]. Among individuals of Caucasian origin, B8 is mainly associated with patients in Northern Europe, in particular in Britain, while B15 is more prevalent in patients from Southern Europe. Apparently, individuals carrying both the B8 and B15 serotypes are twice as susceptible to developing T1DM, as those with either the B8 or B15 serotype alone [6, 39, 40]. Regardless of the B8 dominance, Tait et al. have reported that B18 can be found significantly more frequently in Caucasian patients with clinical T1DM than in antibody positive individuals, and that this association exists in the absence of any class II gene contribution (Table 2) [18]. In contrast, B8 and A1 could be observed at more than twice the frequency in antibody positive individuals (p < 0.0001 for both), while no further increase in frequency from antibody positive to T1DM probands could be found. Surprisingly, A1 was even found significantly less frequently (p < 0.05) in patients than in antibody positive individuals (Table 2) [18]. B8 was also reported in an earlier study as being associated with a significantly increased CD4+/CD8+ ratio indicating that B8 may even be involved in the preclinical stage of diabetes, i.e. the emergence of autoantibodies and the adverse immune response against self-antigens [41]. It is not entirely clear whether class I genes contribute in a more direct or indirect way to the activation and proliferation of autoreactive CD4+ T cells. Evidence for a direct role engaged by class I genes arose from the observation that alloreactive CD4+ T cells are capable of recognizing self-peptides loaded on MHC class I epitopes [42, 43]. In support of the indirect role of class I genes, is the finding that their gene products expressed on dendritic cells prime naïve CD8+ T cells that secret IFN-γ and TNF-α to polarize the development of CD4+ cells [44]. These results suggest that MHC class I molecules are not only contributing to disease progression but also to the occurrence of autoimmunity.
Table
1.
Frequencies and reported results of HLA class I serotypes obtained from association studies |
|
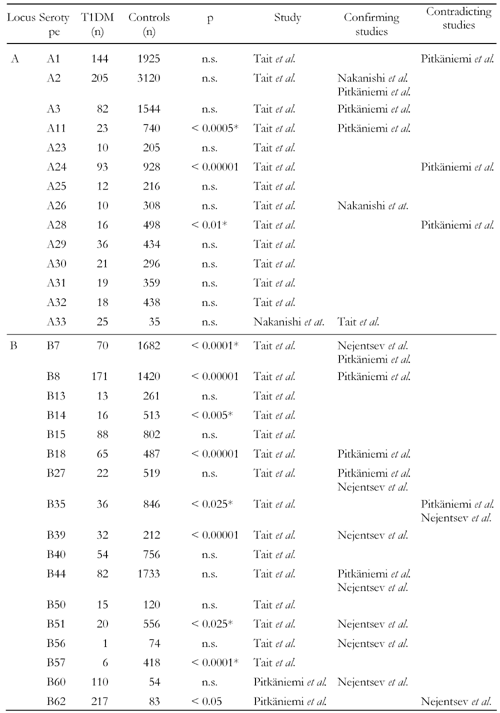 |
 |
Legend:
n: number of subjects in the study. p: reported p-value. * Decreased frequency in T1DM subjects. |
|
Table
2.
Significant serotypes in control, antibody-positive and T1DM individuals detected by Tait et al. |
|
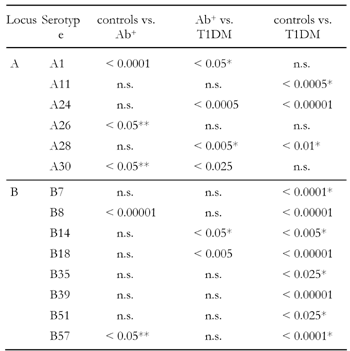 |
|
Legend:
Data represent p-values. Ab+: antibody-positive. * Decreased frequency in T1DM group. ** Decreased frequency in Ab+ group. |
|
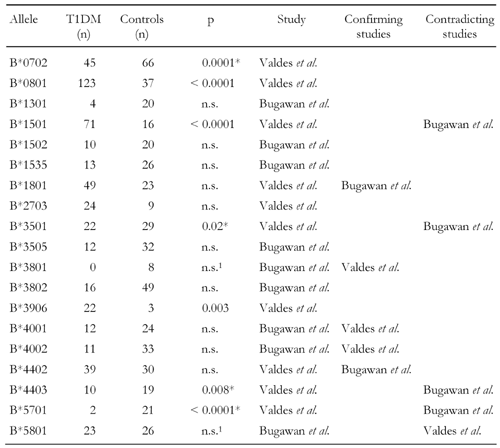
Linkage disequilibrium (LD) is known to exist across the HLA system. This region contains a multitude of genetic loci, which are considered to be jointly responsible for different stages of T1DM development in the context of various degrees of LD with other HLA genes [1]. The identification of genetic linkages to T1DM are thus more difficult given the polygenic character of the disease and the interaction of genetic and environmental factors. Fortunately, improved methods for detecting genetic linkages and hereditary patterns within the HLA system together with modern DNA-based genotyping have led to the identification of more detailed variations of susceptible class I genes and the existence of specific LD patterns [45-48]. In this regard, the early discovery of a significantly increased frequency of the B8 serotype in T1DM probands was detected predominantly in individuals carrying the high risk DRB1*03 allele [18]. B8 was also found to exist more frequently in association with the 8.1 ancestral haplotype, which consists of HLA-A1, C7, B8, C4AQ0, C4B1, DR3, DQ2, including the high risk DR3 serotype, in T1DM patients [49]. An increased CD4+ and decreased CD8+ T cell occurrence as well as increased antibody titers have been described in association with this haplotype [41], which is consistent with the result derived by Tait et al. [18] and appears to be attributed to the A1 and B8 participation (Table 1). The B39 serotype is reported to exist with increased frequency in DRB1*0404-DQB1*0302-positive diabetic patients of Eastern European and Russian origin (p < 0.0001) [50]. Allele B*5801 was reported as being positively associated with disease only in the Filipino but not in another Caucasian cohort (Table 4) [12, 51].
Table
3.
Frequencies and reported results of HLA-A alleles in association studies |
|
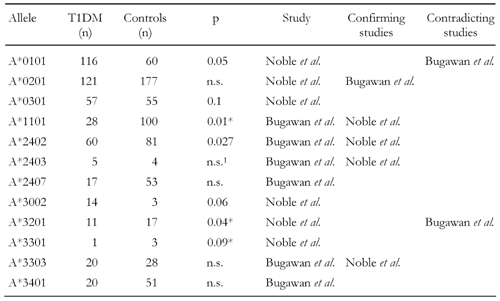 |
|
Legend:
n: number of subjects in the (separate) study. p: reported p-value. * Decreased frequency in T1DM subjects. 1 Significance of A*2403 was calculated in the study in combination with A*2402 yielding p = 0.008. Separately tested: not significant. |
|
Table
4.
Frequencies and reported results of HLA-B alleles in association studies |
|
 |
|
Legend:
n: number of subjects in the study. p: reported p-value. * Decreased frequency in T1DM subjects. 1 Reported as slight association with T1DM, but corrected p-value not significant. |
|
Studies conducted with HLA-A locus antigens have confirmed that the A24 serotype is predominantly existent in antibody positive individuals [18, 21, 26, 27, 52] (Table 2). It is simultaneously associated with a young age of onset [11, 26, 53] and with almost complete loss of residual β-cell function [26, 52]. The relevance of A24 in T1DM was further confirmed in recent studies by Noble and co-workers in a Caucasian (p = 0.042) as well as by Bugawan et al. in a Filipino cohort (p = 0.027) demonstrating allele A*2402 being existent at more than twice the frequency in patients than controls (Table 3) and being strongly associated with total β-cell destruction [11, 26, 51, 54]. The study by Noble et al. conducted with family-based association controls and known LD associations from CEPH families (Centre d'Etude du Polymorphisme Humain) revealed that A*2402 exists at a higher frequency in Caucasian diabetics in the absence of LD with several class II DR and DQ risk conferring haplotypes [54]. Other HLA-A locus alleles, such as A*3002 and A*0101, are also identified as being susceptible in promoting disease development. Their association with T1DM appears to have more of a secondary nature to disease due to LD with the high risk DRB1*0301-DQB1*02 haplotype found in T1DM patients of Caucasian origin [54]. However, several alleles, such as A*2403, A*3002, A*3301, B*2703, B*3801, B*5701 and C*0802, that were reported as being negatively or positively associated with disease, exist rather infrequently (Tables 3, 4, 5). The frequency of these alleles in the two populations varies between 0 and 4.3% [51, 54]. Therefore, their appearance cannot be regarded as representative and the results must be interpreted carefully. Given the rarity of appearance, it is instructive to merge the frequencies of these alleles in a combined group in order to avoid selection biases, as was done in the cross-study analysis. The results of this analysis are presented in the latter section of this article.
Table
5.
Frequencies and reported results of HLA-C alleles in association studies |
|
 |
|
Legend:
n: number of subjects in the study. p: reported p-value. * Decreased frequency in T1DM subjects. |
|
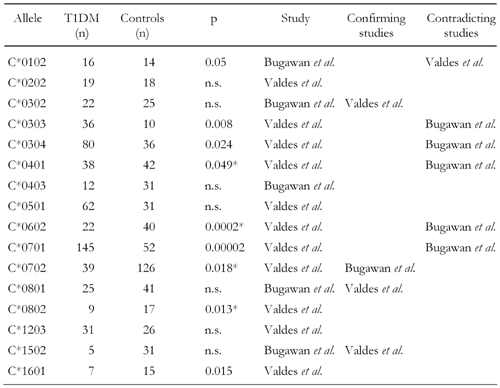
The involvement of genes at the HLA-B and C loci in T1DM development has only recently been explored. In the Caucasian cohort, alleles B*0801, B*1501, C*0303, C*0304 and C*0701 were positively associated with T1DM (Table 4 and 5) [54]. Among these susceptible alleles C*0302 is reported to be in LD with the high risk class II allele DRB1*0301. Therefore, its direct contribution to diabetes progression is unclear, it may at most be accounted to serve as a subordinate marker for the disease. C*0102 has not been found to be in LD with another class II allele or haplotype suggesting it has a more direct role in disease development.
Negative associations of HLA class I genes with T1DM
Linkage analysis has revealed that particular genes within all three HLA class I loci (A, B and C) can be significantly decreased in antibody positive individuals and patients. While serotypes A26 and A30 were found at decreased frequencies in individuals in the stage of progression to overt T1DM compared to controls, A1 appeared to occur negatively associated with overt disease but not with the antibody positive state (Table 2) [18]. The same study reveals that A11 and A28 are decreased in patients vs. controls (Table 1). Among the Filipino patients, the frequency of the A*1101 allele was found to be negatively associated with T1DM, which was in LD with both protective and susceptible haplotypes, DRB1*0803-DQB1*0601 and DRB1*0901-DQB1*0303, respectively [51]. A*2407 was more frequently found in healthy individuals and existed in weak LD with the neutral/weakly protective alleles DRB1*1101 and DRB1*1202 [51].
Serotypes and alleles negatively associated with disease are also present in the HLA-B and C loci. For the B locus, B7, B14, B35, B51 and B57 serotypes have been reported to be present at lower frequencies in T1DM probands compared to non-diabetics (Table 1) [18]. Linkage analysis has further revealed that several HLA-B and C alleles may confer protection. In this regard, B*1301 was found more frequently in healthy individuals in the Filipino population [51]. However, only 7.3% of this population in the analyzed sample carry this allele. Due to this rarity, no robust test statistic can be expected and thus the involvement of this allele must be regarded as undependable. The negative associations of B*0702 and B*3501 reported to occur in a Caucasian population seem to be more robust due to significance and the higher numbers of occurrence (Table 4) [54]. Within the HLA-C locus, C*0702 is reported to appear significantly more frequently in healthy persons than in patients [51, 54], but this association was observed to be in LD with the protective DRB1*1502 allele [51]. In contrast, C*0602 was significantly decreased in patients only in the Caucasian but occurred more frequently in the Filipino cohort (Table 5) [51, 54]. Interestingly, despite the fact that C*1502 is in LD with the risk DRB1*0405 allele, it is still detectable at decreased frequency among the patients examined in the Filipino cohort [51].
Cross-study analysis of HLA class I gene association with T1DM
Aim and background
The incidence of T1DM varies widely among different ethnic groups, as does the distribution of HLA class I and II genes in different populations. Therefore, the analysis of HLA patterns in association with the disease within a pooled population can be instructive in order to identify the most susceptible genetic predispositions by compensating environmental determinants driven by region and ethnology and to enlarge the sample in order to reduce the sample error.
Subjects and data selection
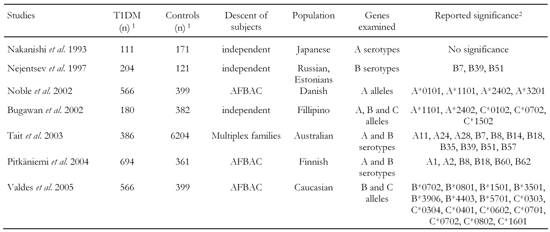
Data was pooled of an overall number of 2707 T1DM and 7638 healthy control haplotypes taken from seven separate studies that compared HLA class I allele occurrence between both these groups (Table 6) [12, 18, 26, 50, 51, 54, 55]. Individuals described in the previous studies were originally typed by sera or DNA samples using polymerase chain reaction. As some studies typed alleles on the basis of carriers (i.e. a single haplotype per carrier) and others on single allele level, carriers of matched characteristics as single haplotypes were regarded in order to yield consistency of the pooled data. No further differentiation of data by genotype or on subject level was considered. The frequencies of the identified characteristics differ from haplotype frequency due to multiple identification of alleles on identical haplotypes. Data was selected carefully by considering any frequencies of clearly identified alleles. Characteristics with infrequent appearance in both subject groups after data pooling were merged into a combined group in order to avoid selection biases.
Table
6.
General parameters and reported results of studies incorporated in the cross-over comparison |
|
 |
|
Legend:
n: number of included haplotypes, AFBAC: affected family-based control. 1 Number of individuals included in the cross-over study (allele number can be lower or higher dependent on occurrence). 2 Either significantly increased or decreased in T1DM group. |
|
The data are heterogeneous regarding ethnology, region and heredity. While controls of some studies are unrelated [26, 50, 51], other studies included affected family-based controls (AFBAC), i.e. either unaffected siblings or children of patients served as control individuals [12, 54]. The antibody-positive group in Tait et al. is pooled by subgroups of related and unrelated affected individuals. The transmission rate of specific DR-DQ haplotypes within the AFBAC can be assumed to be higher than for unrelated controls. Differences in allele frequencies among the studies may thus be caused by ethnology as well as descent dependency, reflected by different haplotype transmission rates. A pooling of independent and descent dependent controls, as conducted in this analysis, balances the overall control group regarding this determinant. AFBAC analysis allows for examining genetic and environmental determinants of a disease, but is also afflicted with some weakness. It is well known that population association between a disease and a genetic marker can arise as an artifact of population structure, even in the absence of linkage to disease. On the other hand, linkage studies without affected sibling pairs may fail to detect linkage if there is a large linkage heterogeneity [56]. Due to the lack of relevant data, haplotype penetrances or LD patterns have not been determined and the control group is considered as being random, i.e. subjects are heterogeneous regarding susceptibility. This leaves an examination of overall risk of alleles and serotypes in randomly pooled individuals of different populations. The aim of this procedure is thus to test if class I alleles identified previously in smaller samples as being at risk of conferring can be confirmed in a larger sample.
Statistical methods
Data were pooled on serotype and single allele level and analyzed per locus and on each level separately in order to yield consistent allele matching. Pearson's χ2-test was applied to determine significance of the comprised HLA alleles and serotypes for each locus. In order to improve approximation, frequencies of characteristics on serotype and allele level were merged that occurred at observed frequencies of less than 10 in at least one subject group, patients or controls. This makes the calculation robust against biases caused by small numbers. Bonferroni correction of p-values (pc) was applied for conservative calculation determined by the number of tests carried out per locus.
Association of class I serotypes with T1DM
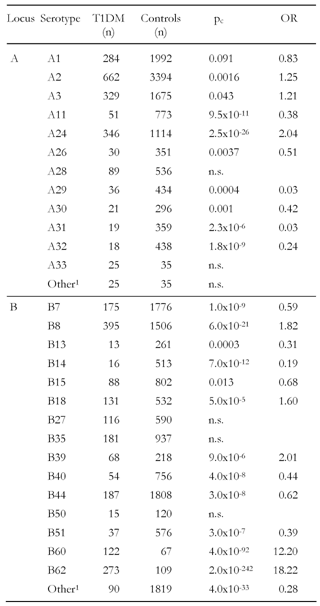
The test for independence of class I serotypes between patients and controls confirmed the previously assumed high incidence of genetic variation. The overall significance for genetic variations in A and B serotypes between both groups of patients and controls was high (p < 0.0001). Among the A locus the analysis confirmed that A24 is positively associated with disease (pc = 1.93x10-27, Table 7), which is consistent with previous results [18, 21] and with observations of the age-related contribution to this serotype to disease onset [11, 53] and the association with complete loss of β-cell function [26]. However, in our enlarged sample, representing a heterogeneous population, A2 and A3 were also significant after correcting p values (Table 7). A2 was also found to be positively associated with the disease in a Finnish cohort (p = 0.05) from the "Childhood Diabetes in Finland" (DiMe) study, a prospective family study carried out between 1986 and 1989 [55]. Fennessey et al. reported an increased occurrence of A2 and A3 on the Cw-B56-DR4-DQ8 haplotype in the same Finnish population [21].
Table
7.
HLA class I serotype frequencies obtained from cross-study data |
|
 |
|
Legend:
n: number of subjects in the cross-over study per characteristic. pc: corrected p-value. OR: odds ratio. 1 Combined characteristics including A34, A43, A66, B37, B38, B41, B45, B47, B49, B52, B53, B55, B58, B70, B75. |
|
A11 was found to be decreased in patients (pc = 7.34x10-12, Table 7) confirming results previously reported [18, 55] but significance has been aggravated compared to these results. A28, which was decreased in Australian T1DM subjects [18], was not significant in our enlarged sample. This result corresponds to Pikaniemi et al. [55]. However, four other serotypes were found (A29, A30, A31 and A32) to be negatively associated with disease (Table 7). Most of these four serotypes were not conspicuous before being protective. A30 was even found to be positively associated with T1DM (p = 0.0001) on the A30-B18-DR3 haplotype [10], but this result may be due to the highly susceptible B18 and DR3 alleles (Table 7) [18, 55].
Beside B18 the susceptibility of B8, B39 and B62 in T1DM development was confirmed, as demonstrated previously [18, 50, 55]. Interestingly, a high significance for an increased occurrence of B60 in diabetic patients was also found (pc = 4x10-92), which was not observed in smaller samples [50, 55]. Moreover, seven serotypes in the B locus were existent with decreased frequency in patients (B7, B13, B14, B15, B40, B44 and B51, Table 7). Although allele B*1301 was observed to be decreased in patients previously, among serotypes only B7, B14 and B51 have been reported in previous studies to be protective, the other types were not found to be significantly decreased in these studies [18, 50, 55]. This suggests that even more genes within the B locus are involved in mediating protection against the disease.
Association of single class I alleles with T1DM
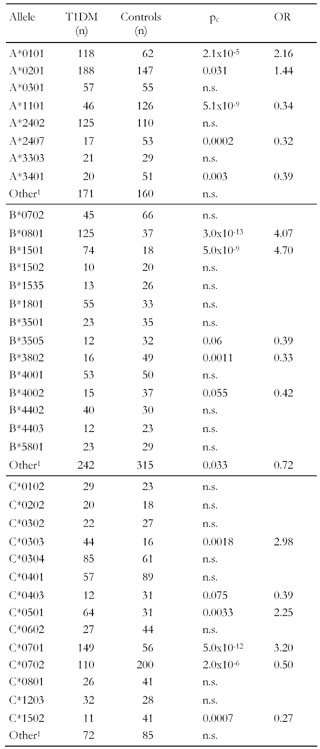
Table 8 shows the frequencies and results from the cross-study on single allele level. Corresponding to previous results, A*1101 is protective. A*3401 is also negatively associated with the disease, which has not been seen in smaller samples. Surprisingly, A*2407 belonging to the A24 serotype family appears to be protective in our enlarged sample. However, this result may be due to the low overall frequency of respective observations in the meta-data. In comparison, the overall frequency of A*2402 has increased after cross-over pooling of data and this allele has become insignificant, suggesting that sample enlargement had a balancing effect on possible selection biases regarding this allele. The converse effect could be observed for allele A*0101, its association with disease increased considerably after data pooling.
Table
8.
HLA class I allele frequencies obtained from cross-study data |
|
 |
|
Legend:
n: number of subjects in the cross-over study per characteristic. pc: corrected p-value. OR: odds ratio. 1 Combined characteristics including A*0203, A*2403, A*2601, A*2902, B*1301, B*1513, B*1521, B*2706, B*4801, B*5101, C*0704. |
|
Two B alleles were found to be strongly associated with disease, B*0801 and B*1501 (pc < 5x10-9, OR > 4, Table 8). While this result confirms the finding of Valdes et al. [12] regarding B*0801, B*1501 appeared to be significant only in the much larger cross-sample. The frequency amounting to (T1DM/controls) = (4/20) in the Filipino population (Table 4) [51] is too small to achieve conventional statistical power, which may explain the different results and emphasize the relevance of both alleles in mediating islet destruction by B8- and B15-restricted CTLs. Three B alleles, B*3505, B*3802 and B*4002, appeared protective for the first time in the cross-over sample. However, this result may be attributed to the frequency of the occurrences that remain small even after data pooling.
The high susceptibility of alleles C*0303 and C*0701 was confirmed in the study (Table 8). In addition, a third allele, C*0501, was identified to be positively associated with the disease. C*0702 was negatively associated analogously to the findings by others [12, 51], but the association appeared much more significant in the large sample analyzed here. C*0403 and C*1502 are also significantly decreased in the patient group, but the occurrence of these alleles is still rare.
Discussion
There are only few studies which hitherto analyzed the association of HLA class I genes with T1DM [12, 18, 26, 50, 51, 54, 55]. Most of them were only recently carried out following the growing insight that class I genes may indeed be essentially involved in the clinicopathology of autoimmune diabetes. Apparently, the more recently a study is conducted, the larger the number of identified alleles associated with the disease (Table 6). However, these studies basically lack statistical power due to the small size of the samples analyzed. Several characteristics existed at a frequency of smaller than ten and these frequencies were used to carry out separate significance tests, which must be regarded as undependable and unbalanced (e.g. Bugawan et al. analyzed 41 HLA-B alleles in separate significance tests including 30 with a frequency of 0-8 just amounting 0-2% of overall appearance). Considering such small numbers would require the application of a simulation-based model, as applied in Pitkaniemi et al. [55]. Therefore in this study, the samples were enhanced by pooling separate samples and matching data exactly on serotype and allele level in order to reduce type 1 error accumulation. The larger sample is more robust against biases due to data selection while it revealed significant associations of several serotypes and alleles with T1DM.
The results presented in this enlarged sample revealed new hitherto unknown associations between HLA class I alleles and autoimmune diabetes. However, they should still be regarded as preliminary since many alleles occurred infrequently even after combining data of the seven existing studies. A larger sample would provide more power to identify markers more reliably. It would thus be instructive to carry out further studies. An alternative approach would be to test the data in a simulation model in order to obtain unbiased estimates.
Conclusion
In the process of preclinical T1DM conversion to overt clinical disease, it is commonly assumed that the final destruction of pancreatic β-cells is mediated by the occurrence of diabetogenic CD8+ T cells. Recent linkage analysis has revealed that HLA class I alleles may contribute decisively to T1DM development dependently as well as independently of LD with other HLA class II alleles or haplotypes. Progression of disease seems to be largely influenced by alleles of the A24 and B18 superfamily [18]. A24 alleles were also observed at significantly increased frequency to participate in complete loss of β-cell function and disease onset at early ages [11, 26, 52]. The results of this cross-study obtained from an enlarged heterogeneous sample, suggested that only A*2407 is associated with T1DM but not A*2402. Family-based studies carried out to identify class I alleles which are preferentially transmitted to healthy family members have revealed that such heterogeneity applies for several alleles, such as those of the A11, A28 and B7 subgroups. Generally, this analysis clearly reveals that many serotype families may be involved in the clinicopathology of T1DM, but that a more detailed review is necessary to map detailed allele susceptibilities. Those studies, aimed at the location of class I-restricted disease markers, were only recently carried out. More studies are necessary to test genetic dependencies on the basis of larger samples which would increase statistical power. An accurate definition of disease susceptible alleles will improve our understanding of antigen presentation mechanisms prevailing in the etiology of autoimmune diabetes. This knowledge is necessary for the design of improved immune intervention strategies to halt T1DM progression in patients at risk of developing the disease or those who are already suffering from it.
Acknowledgments:
This work was supported by grants from the SBDR.
References
- Todd JA. Genetic analysis of type 1 diabetes using whole genome approaches. Proc Natl Acad Sci U S A 1995. 92(19):8560-8565. [DOD]
- Aitman TJ, Todd JA. Molecular genetics of diabetes mellitus. Baillieres Clin Endocrinol Metab 1995. 9(3):631-656. [DOD]
- Deng GY, Muir A, Maclaren NK, She JX. Association of LMP2 and LMP7 genes within the major histocompatibility complex with insulin-dependent diabetes mellitus: population and family studies. Am J Hum Genet 1995. 56(2):528-534. [DOD]
- Sia C, Weinem M. Genetic susceptibility to type 1 diabetes in the intracellular pathway of antigen processing - a subject review and cross-study comparison. Rev Diabetic Stud 2005. 2:40-52. [DOD] [CrossRef]
- Gambelunghe G, Ghaderi M, Cosentino A, Falorni A, Brunetti P, Falorni A, Sanjeevi CB. Association of MHC class I chain-related A (MIC-A) gene polymorphism with type 1 diabetes. Diabetologia 2000. 43(4):507-514. [DOD] [CrossRef]
- Green A, Svejgaard A, Platz P, Ryder LP, Jakobsen BK, Morton NE, MacLean CJ. The genetic susceptibility to insulin-dependent diabetes mellitus: combined segregation and linkage analysis. Genet Epidemiol 1985. 2(1):1-15. [DOD] [CrossRef]
- Chang YW, Lam KS, Hawkins BR. Strong association between DQA1/DQB1 genotype and early-onset IDDM in Chinese: the association is with alleles rather than specific residues. Eur J Immunogenet 1998. 25(4):273-280. [DOD] [CrossRef]
- Chen BH, Chiang CH, Lin SR, Chao MG, Tsai ST. The influence of age at onset and gender on the HLA-DQA1, DQB1 association in Chinese children with insulin dependent diabetes mellitus. Hum Immunol 1999. 60(11):1131-1137. [DOD] [CrossRef]
- Komulainen J, Kulmala P, Savola K, Lounamaa R, Ilonen J, Reijonen H, Knip M, Akerblom HK. Clinical, autoimmune, and genetic characteristics of very young children with type 1 diabetes. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes Care 1999. 22(12):1950-1955. [DOD]
- Tait BD, Harrison LC, Drummond BP, Stewart V, Varney MD, Honeyman MC. HLA antigens and age at diagnosis of insulin-dependent Diabetes Mellitus. Hum Immunol 1995. 42:116-122. [DOD] [CrossRef]
- Valdes AM, Thomson G, Erlich HA, Noble JA. Association between type 1 diabetes age of onset and HLA among sibling pairs. Diabetes 1999. 48(8):1658-1661. [DOD]
- Valdes AM, Erlich HA, Noble JA. Human leukocyte antigen class I B and C loci contribute to Type 1 Diabetes (T1D) susceptibility and age at T1D onset. Hum Immunol 2005. 66(3):301-313. [DOD] [CrossRef]
- Iafusco D, Prisco F, Stoppoloni O, Accolla RS, Tosi G. HLA-DQ genotype in early onset insulin dependent diabetes mellitus. J Pediatr Endocrinol Metab 1999. 12(6):887-890. [DOD]
- Svejgaard A, Jakobsen BK, Platz P, Ryder LP, Nerup J, Christy M, Borch-Johnsen K, Parving HH, Deckert T, Molsted-Pedersen L, et al. HLA associations in insulin-dependent diabetes: search for heterogeneity in different groups of patients from a homogeneous population. Tissue Antigens 1986. 28(4):237-244. [DOD]
- Yamamoto AM, Deschamps I, Garchon HJ, Roussely H, Moreau N, Beaurain G, Robert JJ, Bach JF. Young age and HLA markers enhance the risk of progression to type 1 diabetes in antibody-positive siblings of diabetic children. J Autoimmun 1998. 11(6):643-650. [DOD] [CrossRef]
- Shield JP, Wadsworth EJ, Baum JD. The genetic contribution to disease pathogenesis in childhood diabetes is greatest in the very young. Diabet Med 1995. 12(5):377-379. [DOD]
- Savola K, Bonifacio E, Sabbah E, Kulmala P, Vahasalo P, Karjalainen J, Tuomilehto-Wolf E, Merilainen J, Akerblom HK, Knip M. IA-2 antibodies--a sensitive marker of IDDM with clinical onset in childhood and adolescence. Childhood Diabetes in Finland Study Group. Diabetologia 1998. 41(4):424-429. [DOD] [CrossRef]
- Tait BD, Colman PG, Morahan G, Marchinovska L, Dore E, Gellert S, Honeyman Mc, Stephen K, Loth A. HLA genes associated with autoimmunity and progression to disease in Type 1 Diabetes. Tissue Antigens 2003. 61(2):146-153. [DOD]
- Fennessy M, Metcalfe K, Hitman GA, Niven M, Biro PA, Tuomilehto J, Tuomilehto-Wolf E. A gene in the HLA class I region contributes to susceptibility to IDDM in the Finnish population. Childhood Diabetes in Finland (DiMe) Study Group. Diabetologia 1994. 37(9):937-944. [DOD]
- Yamanouchi J, Verdaguer J, Han B, Amrani A, Serra P, Santamaria P. Cross-priming of diabetogenic T cells dissociated from CTL-induced shedding of beta cell autoantigens. J Immunol 2003. 171(12):6900-6909. [DOD]
- Yoon JW, Jun HS, Santamaria P. Cellular and molecular mechanisms for the initiation and progression of beta cell destruction resulting from the collaboration between macrophages and T cells. Autoimmunity 1998. 27(2):109-122. [DOD]
- DiLorenzo TP, Serreze DV. The good turned ugly: immunopathogenic basis for diabetogenic CD8+ T cells in NOD mice. Immunol Rev 2005. 204:250-263. [DOD] [CrossRef]
- Nikolic-Zugic J, Carbone FR. Peptide presentation by class-I major histocompatibility complex molecules. Immunol Res 1991. 10:54-65. [DOD]
- Nakanishi K, Kobayashi T, Murase T, Nakatsuji T, Inoko H, Tsuji K, Kosaka K. Association of HLA-A24 with complete beta-cell destruction in IDDM. Diabetes 1993. 42:1086-1096. [DOD]
- Honeyman MC, Harrison LC, Drummond B, Colman PG, Tait BD. Analysis Of families at risk for insulin-dependent diabetes mellitus reveals that HLA antigens influence progression to clinical disease. Mol Med 1995. 1:576-582. [DOD]
- Ono SJ, Issa-Chergui B, Colle E, Guttmann RD, Seemayer TA, Fuks A. IDDM in BB rats. Enhanced MHC class I heavy-chain gene expression in pancreatic islets. Diabetes 1988. 37(10):1411-1418. [DOD]
- Panagiotopoulos C, Qin H, Tan R, Verchere CB. Identification of a beta-cell-specific HLA class I restricted epitope in type 1 diabetes. Diabetes 2003. 52(11):2647-2651. [DOD]
- Panina-Bordignon P, Lang R, van Endert PM, Benazzi E, Felix AM, Pastore RM, Spinas GA, Sinigaglia F. Cytotoxic T cells specific for glutamic acid decarboxylase in autoimmune diabetes. J Exp Med 1995. 181(5):1923-1927. [DOD] [CrossRef]
- Rolandsson O, Hagg E, Janer M, Rutledge E, Gaur LK, Nilsson M, Hallmans G, Lernmark A. High GAD65 autoantibody levels in nondiabetic adults are associated with HLA but not with CTLA-4 or INS VNTR. J Intern Med 2003. 253(4):447-453. [DOD] [CrossRef]
- Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest 1994. 94(5):2125-2129. [DOD]
- Tanigaki N, Fruci D, Chersi A, Falasca G, Tosi R, Butler RH. HLA-A2-binding peptides cross-react not only within the A2 subgroup but also with other HLA-A-locus allelic products. Hum Immunol 1994. 39(3):155-162. [DOD] [CrossRef]
- Kuzushima K, Sun R, van Bleek GM, Vegh Z, Nathenson SG. The role of self peptides in the allogeneic cross-reactivity of CTLs. J Immunol 1995. 155(2):594-601. [DOD]
- Fruci D, Greco G, Vigneti E, Tanigaki N, Butler RH, Tosi R. The peptide-binding specificity of HLA-B27 subtype (B*2705) analyzed by the use of polyalanine model peptides. Hum Immunol 1994. 41(1):34-38. [DOD] [CrossRef]
- Hiemstra HS, Schloot NC, van Veelen PA, Willemen SJ, Franken KL, van Rood JJ, de Vries RR, Chaudhuri A, Behan PO, Drijfhout JW, Roep BO. Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Proc Natl Acad Sci U S A 2001. 98(7):3988-3991. [DOD] [CrossRef]
- Atkinson MA. Molecular mimicry and the pathogenesis of insulin-dependent diabetes mellitus: still just an attractive hypothesis. Ann Med 1997. 29(5):393-399. [DOD]
- Schloot NC, Willemen SJ, Duinkerken G, Drijfhout JW, de Vries RR, Roep BO. Molecular mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Hum Immunol 2001. 62(4):299-309. [DOD] [CrossRef]
- Segurado OG, Arnaiz-Villena A, Wank R, Schendel DJ. The multifactorial nature of MHC-linked susceptibility to insulin-dependent diabetes. Autoimmunity 1993.15(1):85-89. [DOD]
- Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, Leslie RD. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001. 44(3):354-362. [DOD] [CrossRef]
- Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, Ryder LP, Nielsen LS, Thomsen M, Svejgaard A. HLA antigens and diabetes mellitus. Lancet 1974. 2:864-866. [DOD] [CrossRef]
- Svejgaard A, Platz P, Ryder LP. Insulin dependent diabetes mellitus. In: Terasaki PI (Editor), Histocompatibility Testing 1980, UCLA Tissue Typing Laboratory, Los Angeles 1981, pp. 638-656. [DOD]
- McCombs CC, Michalski JP, deShazo R, Bozelka B, Lane JT. Immune abnormalities associated with HLA-B8: lymphocyte subsets and functional correlates. Clin Immunol Immunopathol 1986. 39(1):112-120. [DOD] [CrossRef]
- Kobayashi H, Kimura S, Aoki N, Sato K, Celis E, Katagiri M. Existence of MHC class I-restricted alloreactive CD4+ T cells reacting with peptide transporter-deficient cells. Immunogenetics 2001. 53(8):626-633. [DOD] [CrossRef]
- Boyle LH, Goodall JC, Gaston JS. Major histocompatibility complex class I-restricted alloreactive CD4+ T cells. Immunology 2004. 112(1):54-63. [DOD] [CrossRef]
- Mailliard RB, Egawa S, Cai Q, Kalinska A, Bykovskaya SN, Lotze MT, Kapsenberg ML, Storkus WJ, Kalinski P. Complementary dendritic cell-activating function of CD8+ and CD4+ T cells: helper role of CD8+ T cells in the development of T helper type 1 responses. J Exp Med 2002. 195(4):473-483. [DOD] [CrossRef]
- Saiki RK, Bugawan TL, Horn GT, Mullis KB, Erlich HA. Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes. Nature 1986. 324(6093):163-166. [DOD] [CrossRef]
- Bugawan TL, Apple R, Erlich HA. A method for typing polymorphism at the HLA-A locus using PCR amplification and immobilized oligonucleotide probes. Tissue Antigens 1994. 44(3):137-147. [DOD]
- Risch N. Assessing the role of HLA-linked and unlinked determinants of disease. Am J Hum Genet 1987. 40:1-14. [DOD]
- Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 1995. 11:241-247. [DOD] [CrossRef]
- Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, French M, Mallal S, Christiansen F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev 1999. 167:257-274. [DOD]
- Nejentsev S, Reijonen H, Adojaan B, Kovalchuk L, Schnevs A, Schwartz EI, Akerblom HK, Ilonen J. The effect of HLA-B allele on the IDDM risk defined by DRB1*04 subtypes and DQB1*0302. Diabetes 1997. 46(11):1888-1892. [DOD]
- Bugawan Tl, Klitz W, Alejandrino M, Ching J, Panelo A, Solfelix CM, Petrone A, Buzzetti R, Pozzilli P, Erlich HA. The association of specific HLA class I and II alleles with type 1 diabetics among Filipinos. Tissue Antigens 2002. 59(6):452-469. [DOD] [CrossRef]
- Nakanishi K, Kobayashi T, Inoko H, Tsuji K, Murase T, Kosaka K. Residual beta-cell function and HLA-A24 in IDDM. Markers of glycemic control and subsequent development of diabetic retinopathy. Diabetes 1995. 44:1334-1339. [DOD]
- Fujisawa T, Ikegami H, Kawaguchi Y, Yamato E, Takekawa K, Nakagawa Y, Hamada Y, Ueda H, Shima K, Ogihara T. Class I HLA is associated with age-at onset of IDDM, while class II HLA confers susceptibility to IDDM. Diabetologia 1995. 38:1493-1495. [DOD] [CrossRef]
- Noble JA, Valdes AM, Bugawan TL, Apple RJ, Thomson G, Erlich HA. The HLA Class 1 A locus affects susceptibility to Type 1 diabetics. Hum Immunol 2002. 63(8):657-664. [DOD] [CrossRef]
- Pitkaniemi J, Hakulinen T, Nasanen J, Tuomilehto-Wolf E, Tuomilehto J. Class I and II HLA genes are associated with susceptibility and age at onset in Finnish families with type 1 diabetes. DiMe Study Group. Hum Hered 2004. 57(2):69-79. [DOD] [CrossRef]
- Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 1993. 52(3):506-516. [DOD]
This article has been cited by other articles:

|
Dominant suppression of Addison’s disease associated with HLA-B15
Baker PR, Baschal EE, Fain PR, Nanduri P, Triolo TM, Siebert JC, Armstrong TK, Babu SR, Rewers MJ, Gottlieb PA, Barker JM, Eisenbarth GS
J Clin Endocrinol Metab 2011. 96(7):2154-2162
|
|

|
Genetic deficiency of Itgb2 or ItgaL prevents autoimmune diabetes through distinctly different mechanisms in NOD/LtJ mice
Glawe JD, Patrick DR, Huang M, Sharp CD, Barlow SC, Kevil CG
Diabetes 2009. 58(6):1292-1301
|
|

|
HLA and infectious diseases
Blackwell JM, Jamieson SE, Burgner D
Clin Microbiol Rev 2009. 22(2):370-385
|
|

|
Genome-wide association studies: progress and potential for drug discovery and development
Kingsmore SF, Lindquist IE, Mudge J, Gessler DD, Beavis WD
Nat Rev Drug Discov 2008. 7(3):221-230
|
|

|
Islet cell autoantibodies in African patients with Type 1 and Type 2 diabetes in Dar es Salaam Tanzania: a cross sectional study
Lutale J, Thordarson H, Holm P, Eide G, Vetvik K
J Autoimmune Dis 2007. 4:4
|
|
|


)
)
)
)
)
)
)
)

