Diabetic Perspectives
| Rev Diabet Stud,
2005,
2(3):165-176 |
DOI 10.1900/RDS.2005.2.165 |
Approaches Towards Endogenous Pancreatic Regeneration
Meenal Banerjee, Meghana Kanitkar, Ramesh R. Bhonde
Tissue Engineering and Banking Laboratory, National Centre for Cell Science, Ganeshkhind, Pune-411007, India.
Address correspondence to: Ramesh R. Bhonde, e-mail: rrbhonde@nccs.res.in.
Keywords: diabetes, pancreatic regeneration, biomolecules, adult stem cells, replication
Abstract
The phenomenon of pancreatic regeneration in mammals has been well documented. It has been shown that pancreatic tissue is able to regenerate in several species of mammal after surgical insult. This tissue is also known to have the potential to maintain or increase its β-cell mass in response to metabolic demands during pregnancy and obesity. Since deficiency in β-cell mass is the hallmark of most forms of diabetes, it is worthwhile understanding pancreatic regeneration in the context of this disease. With this view in mind, this article aims to discuss the potential use in clinical strategies of knowledge that we obtained from studies carried out in animal models of diabetes. Approaches to achieve this goal involve the use of biomolecules, adult stem cells and gene therapy. Various molecules, such as glucagon-like peptide-1, β-cellulin, nicotinamide, gastrin, epidermal growth factor-1 and thyroid hormone, play major roles in the initiation of endogenous islet regeneration in diabetes. The most accepted hypothesis is that these molecules stimulate islet precursor cells to undergo neogenesis or to induce replication of existing β-cells, emphasizing the importance of pancreas-resident stem/progenitor cells in islet regeneration. Moreover, the potential of adult stem cell population from bone marrow, umbilical cord blood, liver, spleen, or amniotic membrane, is also discussed with regard to their potential to induce pancreatic regeneration.
Introduction
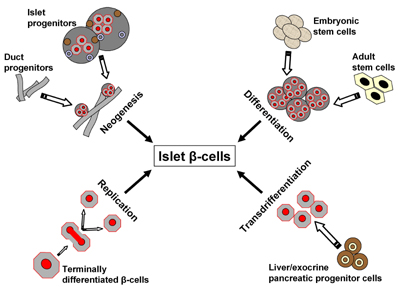
Under normal healthy conditions, organisms maintain a dynamic β-cell mass throughout life, ensuring a sufficient insulin secretion capacity. Critical β-cell mass is maintained as a result of three general mechanisms: (1) replication of existing mature β-cells (2) differentiation (or neogenesis) by ductal or intra-islet pancreatic precursor cells and (3) apoptosis of existing β-cells. An understanding of the critical factors and mechanisms involved in the maintenance of a functional and sufficiently large β-cell mass may be of great value for developing more effective therapies to improve metabolic control or even obtain a cure for diabetes. Many studies have been conducted within the last decade to examine in vitro and/or ex vivo expansion of islets by focusing on phenomena, such as differentiation, trans-differentiation, neogenesis and regeneration, which are illustrated in Figure 1. Several surgical and chemical stimuli have demonstrated islet regeneration. Biomolecules, such as β-cellulin, a member of the epidermal growth factor (EGF) family, are known to trigger in vitro and in vivo replication or regeneration of islets from intra-islet or ductal non-pancreatic stem cells. During pancreatic development, stem cells have a central role in generating endocrine, acinar and duct cells [1]. Recently, the potential of adult stem cells for islet regeneration has been explored. Amongst the various therapeutic approaches, cell therapy is promising due to its possible curative potential. In this review, we discuss the role of potential candidates capable of effecting endogenous islet regeneration along with their modes of action, and we highlight recent developments in this field.
 |
 |
Figure 1. Possible mechanisms of islet β-cell birth. New β-cells may emerge after injury, apoptosis as a result of inflammation, immune-overreactivity or oxidation, or by exogenous stimuli emanating from different cell sources. Neogenesis may originate from progenitor cells residing within the pancreas. Differentiation may be brought about by different subsets of stem cells. Replication occurs when differentiated pancreatic cells duplicate while transdifferentiation is a process thought to be equipped by undifferentiated progenitors not necessarily preassigned to the pancreas. |
|
Role of biomolecules in β-cell regeneration
Biomolecules are active agonists that are synthesized in a biological system. These molecules exhibit tissue specificity, as well as autocrine and paracrine effects, on various cell populations. This may explain their ability to effectuate regeneration in intra- and extra-pancreatic environments. It is certainly desirable to restore damaged tissue by stimulating the regeneration of competent cells. Therefore, the use of biomolecules is an attractive option for β-cell regeneration.
It was shown recently that the exon1 Cys7Gly polymorphism within the β-cellulin gene is associated with type 2 diabetes in African Americans [2]. In the context of this finding, Huotari and colleagues have suggested a role for β-cellulin in islet neogenesis and regeneration [3]. A combination of activin A and β-cellulin, when tested on neonatal streptozotocin (STZ)-treated rats, resulted in the regeneration of pancreatic β-cells and improved glucose metabolism [4]. Furthermore, Li et al. showed that β-cellulin induces differentiation of intra-islet precursor cells (mainly from somatostatin-positive islet cells) into β-cells in STZ-treated mice, and causes a reduction in hyperglycemia [5].
Biomolecules also seem to have a direct effect on the regulation of regeneration and the development of specific genes. Recently, Yoshikumi et al. [6] identified a novel mitochondrial transporter, citrate transporter protein-like protein (CTPL) and its mouse orthologous Sfxn3, a member of the sideroflexin family, which were seen to be up-regulated in STZ-induced diabetic rats in the early phase of differentiation into insulin-secreting cells. The function of some biomolecules from the epidermal growth factor (EGF) family of proteins, e.g. β-cellulin and activin A, during pancreas development is well documented. β-cellulin and activin A seem to be responsible for the up-regulation of these genes in STZ-induced diabetes.
An alternative strategy to treat diabetes is the use of various plant extracts and herbal biomolecules, due to their hypoglycemic effects. Detailed investigation into the modus operendi of these biomolecules has revealed that some of them cause a regeneration of β-cells, thus causing a reversal of diabetes in human and non-human subjects. For example, chard extract (Beta vulgaris L. var. cicla) has been used as a hypoglycemic agent by diabetic patients in Turkey [7]. It could also be observed that the number of β-cells and secretory granules increased in diabetic patients who received this herbal extract. The authors concluded that the extract is able to reduce blood glucose by regenerating β-cells. Similarly, the effects of crude Epherdrae herba, an alkaloid of epherdrae and 1-ephedrine (a major alkaloid component) promoted islet regeneration following atrophy induced by STZ [8]. Another promising agent, nicotinamide, has been shown to protect islets from inflammation and to stimulate endocrine differentiation of rat β-cells in vivo. A dose of 3g/day of nicotinamide was administered to humans for more than a year without any serious side-effects [9]. A stable and functional regeneration of pancreatic β-cells was obtained in neonatal STZ-treated rats that were administered sodium tungstate via drinking water [10]. Recently, Kim et al. have showed an up-regulation of clusterin expression after islet injury, suggesting that clusterin might be involved in cytoprotection and the regeneration of islet cells [11]. Since clinical and epidemiological studies have suggested that reduced zinc status is associated with diabetes, investigators have searched for its role in pancreatic regeneration. A significant correlation has been observed between zinc concentration in pancreatic tissue and the pancreatic regeneration rate [12]. Finally, zinc supplementation was shown to be effective in preventing and ameliorating diabetes in several animal models of type 1 and type 2 diabetes [13]. However, many issues need to be clarified, such as dose determination and side effects, before these agents may be considered for therapy.
Relevance of hormones in β-cell regeneration
The processes associated with pancreatic development and regeneration are mostly controlled by growth-inducing (GI) hormones. One such hormone, cholecytokinin, has been extensively studied for its role in pancreatic regeneration. The infusion of cholecytokinin octapeptide (CCK-8) into rats at a dose of 600ng/kg/hour provoked an acceleration of the regenerative process [14]. GRP-bombesin, another GI neuropeptide hormone, exerts marked growth-promoting action in rodent pancreas [15]. Recently, Fiorucci and colleagues discovered that bombesin induced pancreatic regeneration in pigs mediated by p46Shc/p52Shc and p42/p44 mitogen-activated protein kinase up-regulation [16]. Beside GI neuropeptides, there is another group of hormones, the thyroid hormones, which elicit diverse cellular and metabolic effects in various organs, including mitogenesis in rat liver. Experiments were carried out to investigate whether this group of hormones is able to stimulate cellular proliferation in other quiescent organs, such as the pancreas. In vivo treatment with the thyroid hormone T3 demonstrated that this induced extensive cell proliferation in the exocrine pancreas [17].
Along with β-cellulin, EGF is known to elicit a growth and nesidioblastotic response in the pancreas. It is thus assumed that physiologically significant improvements in glucose tolerance can be achieved by stimulating β-cell regeneration with gastrin/EGF administered systemically as a transgenic expression of gastrin and EGF receptor ligands [18]. Such "hormonal treatments", if proven non-deleterious in other metabolic processes, could be considered as a feasible strategy for the induction of β-cell growth and regeneration in humans.
Interplay between biomolecules and endogenous islet regeneration in pancreatic injury model
The neogenesis of endocrine islets initiated by the ductal epithelium has been described in vivo in various experimental settings and in clinical pathologies. Experiments in models of partially or subtotally pancreatectomized animals have demonstrated that injury is a trigger for tissue repair, thus leading to a kind of self-healing and possibly normoglycemia. It is remarkable that the body appears to be capable of restoring almost full organs as long as there is some organ tissue left from which regeneration can emanate. Intercalated ductal cells and centro-acinar cells are speculated to be the major sources of neogenesis, as was deduced from a study involving 90% pancreatectomized animals [19]. Thus, organ regeneration and the restoration of normoglycemia are possible only upon achieving extremely high levels of replication and/or regeneration of β-cells to 'fill up' the lost cell mass and volume. This dimension of cell mass renewal appears to be impossible if undertaken by one cell mass renewal process alone. In a partial pancreatectomy rat model of type 2 diabetes, the daily administration of exendin 4 stimulates pancreatic regeneration and β-cell expansion by two processes: neogenesis by progenitor cells, and proliferation or replication of mature β-cells [20].
A role for endogenous glucagon-like peptide-1 (GLP-1) has been suggested in β-cell regeneration after 70% partial pancreatectomy. GLP-1, in combination with exendin-4, expands β-cell mass by stimulating neogenesis and proliferation [21]. Insulin receptor substrate-2 (IRS-2) is known to play an important role in pancreatic regeneration and growth by mediating duct proliferation and maintenance of the differentiated β-cells [22]. Agents such as exendin-4, GLP-1 and IRS-2 are known to stimulate regeneration emanating from the existing precursor cell mass and replication of the existing, albeit depleted, β-cell mass; thus, both processes work together in organ regeneration. IRS-2 also adopts the important role of initiating duct proliferation (i.e. increasing the relative number of ductal precursor cells) and the maintenance of differentiated β-cell mass.
Several growth factors have been examined in the model of 90% pancreatectomized animals. Firstly, the expression of insulin-like growth factor-1 (IGF-1), which is known to have varied effects, such as growth promotion, insulin-like influence on glucose metabolism, and neuroprotection resulting from cell-prolifer-ative and antiapoptotic properties, could be observed after 90% pancreatectomy, suggesting that this factor plays a critical role in regulating the regeneration and/or replication of β-cells [23]. β-cellulin (0.5 μg/g body weight) is another factor that was shown to improve glucose metabolism by promoting β-cell regeneration in 90% pancreatectomized rats [24]. In the same model, rats treated with poly (ADP-ribose) polymerase inhibitors (nicotinamide and 3 amino-benzamide) showed islet β-cell regeneration along with expression of the Reg II gene [25]. The latter is assumed to induce regeneration in injured tissue, as it prevented diabetes in other experimental animal models [26, 27].
Duct ligation is another model for the study of islet neogenesis. Duct ligation in fetal and adult pancreas induces islet neogenesis due to an up-regulation of netrin-1, a diffusible laminin-like protein, which is known to up-regulate neuronal cell migration via a chemokinetic effect in the pancreas. Netrin-1 expression was observed in regenerating exocrine and endocrine pancreatic tissue [28]. Rosenberg et al. have shown that, through partial obstruction of the hamster pancreas, STZ-induced diabetes could be reversed in more than 50% of the cases [29]. An extract termed as ilotropin, prepared from obstructed pancreata, seemed to reverse the disease. Ilotropin contains a protein that is capable of stimulating the proliferation of isolated duct cells in culture. Islet neogenesis-associated protein (INGAP) was expressed early in the neogenic process before the onset of ductal cell proliferation, suggesting that this peptide was involved in the initiation of the regenerative process [29].
Local injury induces a complex orchestrated response to stimulate the healing of injured tissues, cellular regeneration and phagocytosis. Our own study has demonstrated that the induction of a regenerative stimulus (pancreatectomy) in conditions of STZ-induced diabetes is indeed able to trigger the pancreatic regenerative process, thereby restoring a functional pancreas [30]. Our experiments in this area of research have led the hypothesis that factors observed during the process of regeneration carry the potential to induce islet neogenesis and normoglycemia. Upon injecting cytosolic extract of regenerating pancreas into STZ-diabetic animals, these animals returned to normoglycemia [31]. Hence, we hypothesized that culture from pancreatic cells is also capable of provoking in vitro regeneration starting from intra-islet precursor cells and leading to the restoration of sustained normoglycemia after injection of the extract into STZ-diabetic mice [32]. This indicated that factors inducing regeneration were secreted into the culture medium upon local injury in terms of islet isolation. A more extensive analysis to identify these factors and their exact functions may one day allow us to harness this strategy for the induction of islet neogenesis. It is evident from the wealth of literature available that there is no shortage of biomolecules that may be able to bring about pancreatic regeneration and, thus, restore normoglycemia. Although this perspective appears extremely positive, it remains highly experimental so far. Most of these biomolecules have been investigated in animal studies, yet they have not reached the stage of clinical trials. Further extensive studies must be carried out before they can be safely extrapolated to humans. It is prudent, therefore, to look at further avenues that may potentially lead to the goal of β-cell self-replication and/or regeneration.
Contribution of β-cell self-replication and regeneration for the maintenance of a critical pancreatic cell mass
We know that regeneration processes are needed to maintain the critical cell mass which is necessary to maintain organ homeostasis. Each cell in the body, even after terminal differentiation and commitment to a particular lineage, preserves a kind of replicative capacity. This capacity of replication or self-duplication allows cells to survive and save their particular functions for several cycles. The potential of replication is limited in the case of pancreatic β-cells. However, in a population of normal adult pancreatic islet cells, the number of β-cells actually undergoing cell division is small, measured to be between 0.5 and 2 % [33]. In normal scenarios with limited apoptosis and natural cell death, organs are able to maintain homeostasis, even if the cellular component of one of their tissues has a restricted replication rate. Imagine a situation of infection or disease with an accelerated damage or death of β-cells. Then the question arises as to which forces come into play in order to augment the cell mass and to restore the full organ function. Only in recent years has it been revealed that the pancreas can indeed undergo repair following damage, a process known today as regeneration. Several findings have now proved that β-cells, although they do not exhibit a particularly high replicative potential, are able to regenerate their own cell mass under disease conditions. In this context, Asanuma et al. [34] proposed a transplantation model to study the in vivo regeneration machinery of the pancreas and liver, where functional cells were transplanted directly into these organs. This report established the fact that, similar to the liver, the fetal and the adult pancreas is a regenerative competent organ. A study by Yamamoto et al. showed that the fetal pancreas treated with STZ has the ability to regenerate β-cells in vivo [35]. Their morphometric studies of STZ-treated diabetic rats demonstrated that the increase in β-cell mass was not due to β-cell hypertrophy but to hyperplasia, accompanied by an increase in islet density in the treatment group of rats. Once it was established that pancreas can regenerate, researchers started to look at the factors and stimuli that instruct pancreatic tissue to undergo regeneration. Investigators have examined both pancreatic as well as non-pancreatic proteins that notionally possess islet neogenesis potential. Therefore, it seemed obvious to test the islet neogenesis potential of insulin, the pancreas' own secretory protein. In their study, Guz et al. tested whether the restoration of normoglycemia by exogenously administered insulin can enhance β-cell differentiation and maturation [36]. They found that β-cell regeneration improved in STZ-treated mice. Thus, they concluded that β-cell neogenesis occurred in adult islets and is regulated by insulin-mediated normalization of circulating blood glucose levels. Furthermore, Brand et al. observed that treatment with gastrin, together with EGF, both of which are non-pancreatic proteins, stimulated β-cell regeneration in chronic insulin-dependent diabetes [12]. The EGF/gastrin-treated group exhibited a significantly greater number of BrdU labeled β-cells, which indicated the stimulation of β-cell replication or neogenesis. Thus, their study showed that physiologically significant improvement in glucose tolerance can be achieved through stimulating β-cell regeneration with gastrin/EGF as a conventional pharmacological therapy.
Do pancreas-resident stem cells exist?
Of crucial importance in the development of diabetes, both in type 1 and type 2, is the insufficient β-cell replication after the onset of disease. This is why we are always in search of new sources of β-cells to be generated by neogenesis or for factors to induce endogenous β-cell replication. In this regard, pancreatic stem or progenitor cells may offer a promising therapeutic approach for diabetes. It is clear that pancreatic progenitor cells exist and play a major role during the embryonic development of the pancreas, as well as in adult life, evidenced by the study of Guz et al. which was successful in identifying two presumptive precursor cell types that appear in regenerating islets [36]. One cell type expressed the glucose transporter-2 (Glut-2), and the other one co-expressed insulin and somatostatin. Although further intensive studies need to be carried out to learn about the mechanisms and molecules that control these cells and processes, an elegant genetic lineage study demonstrated recently that, following partial pancreatectomy to trigger regeneration, the replication of existing β-cells is a dominant pathway for formation of new β-cells in adult mice [37]. Other than studies assessing cell position, gene and/or protein expression and proliferative states, which can change rapidly, Dor and colleagues used methods to track the fate of islet β-cells. They used a genetic method to indelibly mark the DNA of adult mouse β-cells, such that they could definitively track the cells and their descendants during normal and stimulated cell turnover. The authors added a hybrid gene into mice that contained the insulin promoter linked to a gene for a recombinase, an enzyme that is able to rearrange specific DNA sequences in a cell. The insulin regulatory sequence caused the recombinase to be expressed only in β-cells and not in duct cells or other non-β-cell types. An additional twist was that the recombinase was inactive unless the animal was treated with a synthetic hormone. When Dor et al. administered a pulse of this hormone to the mice, it activated the recombinase, which led to a labeling of the cells by genetic rearrangement. After the hormone pulse, all of these marked cells examined were insulin-positive β-cells. In a second step, the authors tracked descendants of the marked cells for up to a year and determined their fate during both normal cell turnover and after partial pancreatectomy. Surprisingly, the authors found that the percentages of labeled β-cells, during both normal turnover and pancreatic regeneration, remained stable and were not superseded by β-cells that could have arisen from insulin-negative progenitors (duct or stem cells). In other words, many of the β-cells generated over the study period were labeled cells, indicating that they originated from cells that expressed the recombinase at the time the hormone pulse was given and these cells were β-cells. The authors' quantification showed that the contribution of non-β-cells (duct or stem cells) was minimal. They also failed to find new, unmarked islets during normal β-cell turnover; such unmarked islets would be predicted if other cell types could generate β-cells.
Another study demonstrated clonal derivation of multipotent precursor cells of both islet and ductal origin [38]. All major islet endocrine cell types could be derived from these precursors, even if the insulin-producing cells cannot be considered bona fide, fully differentiated β-cells at this stage. How can the apparent conflict between these two studies be resolved? A fundamental difference of obvious significance for regenerative or cell-based therapy in diabetes is that the first was performed in vivo [37] and the second in vitro [38].
A substantial amount of evidence has accumulated over the last ten years supporting the hypothesis that islet neogenesis in the mature pancreas occurs via cells which are located in, or which are associated with, the ductal epithelium. These cells have been phenotypically defined in human and animal models through expression patterns of specific cytokeratin intermediate filament proteins. It has thus become increasingly evident that early pancreatic development from the endodermal bud progresses via a kind of "branching morphogenesis" of cytokeratin-expressing ductal structures [39, 40] and that cells within these structures possibly lose cytokeratin expression and develop into both the endocrine and exocrine compartments [40]. Similarly, in the adult pancreas, individual β-cells, as well as intact islets, have been observed in close contact with the cytokeratin-positive ductal epithelium [41, 42]. In addition, the same authors described the appearance of transitional cells expressing both insulin and CK19 in adult human pancreatic sections [42]. These data suggest that pancreatic stem cells appear to be of ductal origin or to reside in close association with the ductal epithelium.
Data from both in vivo and in vitro experiments support the hypothesis that islet neogenesis in the mature pancreas emanates from ductal cells. In 1991, it was shown that the co-transplantation of adult rat pancreatic epithelium and fetal-derived mesenchyme into the epididymal fat pads of rats resulted in the appearance of cells expressing islet hormones [43]. Furthermore, Bonner-Weir et al. [44] cultured human ductal cells as a monolayer overlaid with Matrigel™, and observed the formation of "islet buds" containing CK19-expressing cells, as well as insulin-positive cells. Although these studies showed convincing evidence that ductal cells contribute to islet neogenesis in the adult pancreas in vitro, the low proportions of differentiating cells suggested that either the methods were as yet inefficient, or that only a specific sub-population of ductal cells are true islet progenitors.
Contribution of adult stem cells to islet neogenesis
Adult stem cells are often known as tissue-specific stem cells. Since they are tissue-restricted they have a lower plasticity in terms of crossing borders of cells types, as compared to embryonic stem cells. Hematopoietic and mesenchymal stem cells are considered to be the most plastic stem cells as they can not only differentiate into mesodermal cell lineages but may also transdifferentiate to capture the phenotypes of ectodermal and endodermal lineages. Several researchers have shown the potential of bone marrow stem cells to differentiate into pancreatic islet cells. Apart from blood and bone marrow stem cells, other tissue-resident stem/progenitor cell resources have also been demonstrated to have islet neogenesis potential.
Bone marrow stem cells
Bone marrow has tremendous clinical implications, since it is relatively easy to collect bone marrow stem cells for further in vitro manipulations. Bone marrow transplantation has already become a powerful strategy for the treatment of hematologic and metabolic disorders and is also considered to be effective in autoimmune diseases. There is a great debate on the issue of the fate of transplanted bone marrow stem cells, whether they undergo cell fusion or transdifferentiation into pancreatic β-cells, or if they induce endogenous pancreatic regeneration in vivo by stimulating neo-vascularization. Autoimmune diabetes in non-obese diabetic (NOD) mice can be prevented by allogeneic bone marrow transplantation (BMT) from diabetes-resistant murine strains [45]. Hess et al. reported a lowering of blood glucose levels within a week after intravenous infusion of green fluorescence protein (GFP)-tagged allogenic bone marrow into STZ-induced diabetic mice [46]. Only a low frequency of donor-derived insulin-producing cells was found in the host; islets were found to be largely replenished by endogenous cells. The authors suggested that donor GFP-positive bone marrow stem cells "only" guided the endogenous growth of islets. The phenomenon of islet cell derivation from bone marrow stem cells in vivo has also been observed by Ianus et al. [47]. The group showed that bone marrow-derived cells can differentiate into insulin-producing islet cells with glucose-dependent and incretin-enhanced insulin secretion when transplanted into lethally irradiated mice. However, other groups working on bone marrow stem cells have not observed such an anti-diabetic effect after bone marrow infusion [48, 49, 50]. Thus the issue of restoration of pancreatic cell function by bone marrow transplantation still remains debatable.
If we leave aside the discussion regarding the fate of infused bone marrow cells within the pancreas and try to establish a correlation between allogenic chimera formation in the host after bone marrow infusion and the recovery of physiologically adequate endogenous insulin regulation, we find that as little as 1% of the initial allogenic chimerism can reverse the diabetogenic processes in the islets of Langerhans in pre-diabetic NOD mice [51]. The restoration of endogenous β-cell function to physiologically sufficient levels is achievable even though the allogenic bone marrow transplantation is performed after the clinical onset of diabetes. Thus, authors claim that, if the same patterns of islet regeneration would appear in humans, the induction of an autoimmunity-free status by the establishment of a low level of chimerism might become a new therapy for type 1 diabetes. Bone marrow is not the only source of hematopoietic stem cells (HSC), it also includes other stem cells such as mesenchymal stem cells (MSC) and endothelial progenitor cells (EPC). Beside exploring the role of HSC in pancreatic regeneration scenarios, researchers have also explored the islet neogenesis potential of EPC. Recently, Mathews et al. provided evidence that bone marrow-derived EPC could be recruited to the pancreas in response to islet injury [52]. Although bone marrow-derived EPC contribute to neovascularization, they do not differentiate into insulin-expressing cells. Attempts have been made to apply bone marrow transplantation, either by using whole unfractionated bone marrow, or with one of its major stem cell fractions. Our own study showed that unfractionated bone marrow derived from experimental diabetic mice, when transplanted into STZ diabetic mice, contributed significantly to blood glucose improvement. This study also demonstrated that multiple injections of bone marrow restore normoglycemia in experimental diabetic mice by retaining the potential to induce endogenous pancreatic regeneration [53]. We succeeded recently in tracing the path of infused bone marrow cells in vivo, employing LacZ-labeled bone marrow of ROSA26 transgenic mice. Beilhack et al. employed a purified hematopoietic stem cell fraction of the bone marrow to test if the development of hyperglycemia could be prevented in NOD mice [54]. The data demonstrated that the purified HSC grafts block the development of autoimmune diabetes and illuminate how HSC grafts alter thymic and peripheral T cell responses against auto- and allo-antigens. Several studies have conclusively demonstrated that both major stem cell components of the bone marrow (HSC and EPC) have the ability to home to the damaged pancreas to induce endogenous regenerative processes. In this regard, recently Izumida et al. proposed a possible mechanism supporting the recovery of β-cells after bone marrow transplantation [55]. They provided evidence for the involvement of the c-Met/HGF pathway in the recovery of β-cells after injury. Thus, they claim that the induction of the c-Met/HGF-signaling pathway following bone marrow transplantation promotes pancreatic regeneration in diabetic rats. Although these approaches are promising due to their potential to induce regeneration of the diabetic pancreas, more information is needed to fully understand how stem cells mediate endogenous regeneration.
Liver stem cells
The liver is an organ with a tremendous regenerative capacity. Therefore, liver oval cells may be a promising source of islet cells, as they develop in close relation to the pancreas [56]. c-Met+CD49f+/-c-Kit-CD45-TER119- cells have been identified as hepatic stem cells in the murine fetal liver. These cells possess multilineage differentiation potential and self-renewing capability. They can differentiate in vivo into some epithelia capable of reconstituting tissues within the liver, pancreas and intestine following appropriate transplantation [57]. Kojima et al. reported islet neogenesis in the liver and a reversal of diabetes in mice by adenovirus delivery of neuro D and β-cellulin [58].
Intestinal stem cells
The gut contains one of the largest stem cell populations in the body, yet has been largely overlooked as a source of potentially therapeutic cells. The stem cells reside in the crypts located at the base of the protruding villi, reproduce themselves, and repopulate the gut lining as differentiated cells by sloughing off into the lumen [59]. Identity, distribution and regulation of small intestinal epithelial stem cells and their immediate progeny have been discussed by Kodama et al. [60].
Spleen cells
The spleen is an organ that has been disregarded for long. Only few recent studies on this source are available, but they provide evidence of the existence of spleenic stem cells. The studies suggest that the spleen may be a reservoir of HSC. Recent studies where spleenic stem cells were shown to differentiate into cells of other organs, including pancreatic β-cells, have opened up an avenue for the spleen to be a target for cell-based therapy to treat diabetes. Kodama et al. demonstrated that NOD mice with end-stage disease administered with live labeled donor male splenocytes rapidly differentiate into islet and ductal epithelial cells within the pancreas [61]. In addition, this data suggested a role for complete Freund's adjuvant together with the injection of donor splenocytes to eliminate autoimmunity and restore stable normoglycemia. The return of endogenous insulin secretion was accompanied by the reappearance of pancreatic β-cells. Moreover, the recovered islets were persistent and functional. In these experiments, the authors identified a marker, Hox11, expressed by spleen stem cells, a highly conserved transcription factor that plays a major role in the development of organs in vertebrate and invertebrate embryos. These stem cells obviously have the capacity to effectively regenerate into insulin-producing islet cells in diabetic mice or genetically altered mice that lack a pancreas. Although the authors call for reappraisal of the spleenic cells for treating diabetes, it should be noted that no other group has been able to successfully reproduce these studies. Another study describing pancreatic agenesis [62] reported that the endocrine pancreatic cells originate in the spleen. No lineage studies have yet demonstrated the transdifferentiation of spleenic cells into hormone-producing cells of the pancreatic islets of Langerhans.
Cord blood stem cells
Several studies have shown that cord blood stem cells are a source of putative hematopoietic and endothelial stem cells. Researchers have made use of these cord blood-specific HSC and EPC to investigate whether they can play a role in blood glucose lowering by protecting islets in a diseased or damaged pancreas. It was found that intravenous transplantation of a mononuclear cell (MNC) fraction of the cord blood in NOD mice resulted in a significant lowering of blood glucose levels and a significant reduction of insulitis. Further investigations led to the conclusion that these cells actually improve type 1 diabetes by protecting islets in NOD mice [63]. The beneficial effects of cord blood MNC are not restricted to type 1 diabetes, they are also found to be effective in type 2 diabetes. Upon transplantation of human umbilical cord blood MNC in obese mice with spontaneous development of type 2 diabetes, the mice exhibited beneficial effects on blood glucose levels, survival and renal pathology [64]. Not only MNC, but also one of the purified fractions of cord blood, namely EPC, have been shown to improve circulation to ischemic tissues. Naruse et al. examined whether therapeutic neo-vascularization using human umbilical cord blood-derived EPC reverse diabetic neuropathy [65]. Their findings suggest that transplantation of EPC from cord blood may be a useful treatment for diabetic neuropathy. Thus, cord blood stem cells offer another option of stem cell therapy in diabetes mellitus.
Amniotic membrane stem/progenitor cells
The amniotic membrane is part of the fetal membrane and is composed of amniotic epithelium and mesenchymal cells. Since amniotic membrane epithelium and mesenchymal cells are derived from the inner cell mass of the blastocyst, they are expected to possess the potential to differentiate into various cell lineages. In this regard, Wei et al. demonstrated that amniotic membrane epithelial cells are capable of normalizing the blood glucose level of STZ-induced diabetic mice after several weeks of implantation [66]. Hence, these cells have therapeutic potential for the treatment of type 1 diabetes. It is indeed difficult to estimate the ability of this versatile "warehouse" of non-pancreatic stem cells to contribute to the course of a cure for diabetes. However, the presence of such ectopic sites of β-cell neogenesis is suggestive of their possible contribution towards β-cell regeneration. Future investigations of cell therapeutic approaches for provoking endogenous islet regeneration for their contribution would therefore be worthwhile.
Gene therapy and β-cell replication and regeneration
Gene therapy holds a tremendous therapeutic potential to improve glycemic control by restoring endogenous insulin production. Insulin gene therapy is thought to overcome the hurdles of islet transplantation and β-cell regeneration with respect to their vulnerability to autoimmune attack. The major issues when dealing with gene therapy approaches for diabetes refer to the use of a system that would allow for efficient transcription, translation, post-translational modifications, packaging and secretion of mature insulin in a glucose-dependent fashion. Luo et al. investigated whether systemic TGF-β1 gene therapy can block islet-destructing autoimmunity and facilitate the regeneration of β-cell function in overtly diabetic NOD mice [67]. For this purpose NOD mice with established diabetes were transplanted with 500 syngeneic islets under the kidney capsule after 7-14 days of receiving an intravenous injection of Ad-hTGF-β1. The results demonstrated that systemic TGF-β1 gene therapy blocks islet-destructing autoimmunity, facilitates islet regeneration, and cures diabetes in NOD mice. Due to its capacity of activin to regulate the growth and differentiation of a variety of cells, and due to its assumed role in the specialization of pancreatic precursors from the gut endoderm during mid gestation, Zhang et al. tested the role of activin signaling during pancreatic islet cell development and regeneration [68]. Their results documented the up-regulation of both activins and activin receptors in duct epithelial cells during islet differentiation while the inhibition of activins significantly enhanced the expansion of pancreatic epithelial cells but decreased the number of differentiated β-cells. Another study carried out by Chaorui et al. is of clinical significance in the context of islet transplantation and prevention of the recurrence of autoimmunity after transplantation [69]. Their data suggested that the introduction of diabetes-resistant MHC class II β-chain in bone marrow-derived cells is sufficient to prevent the development of functional islet-reactive T cells, and to mediate the intra-thymic deletion of these cells. The expression of diabetes-resistant MHC class II β-chains prevented the development of functional T cells that respond to specific islet self-antigens which are targets of T cell-mediated destruction of pancreatic β-cells. Morral reviewed the recent advances directed at producing insulin in an ectopic tissue, as well as inducing pancreatic β-cell neogenesis [70]. The discussed strategies include constitutive and promoter-regulated insulin expression in the liver; increasing hepatic glucose oxidation, insulin production from intestinal cells and islet cell neogenesis in the liver and the pancreas.
Although the pancreas has the capacity to regenerate, why does it not happen in diabetes?
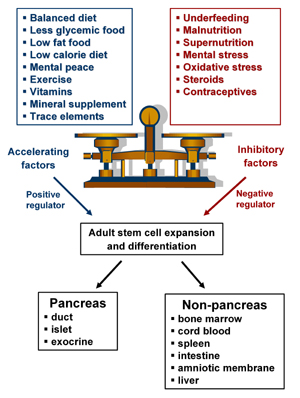
We have discussed so far that the pancreas has regenerative potential in embryonic, neonatal and adult life. However, this potential does not seem to help during progression to diabetes. The diabetic environment, viz hyperglycemia, changes the lipid profile and, coupled with glycemic and high fat diet, precipitates the disease, preventing regenerative processes. Any conditions that may disturb β-cell birth and the death balance result in overt diabetes. The critical balance between exogenous acceleration and inhibitory factors, which maintain cell health in vivo and the imbalance of any of these results in diabetes, are summarized in Figure 2. Most of the physiological processes are coordinated by homeostasis which maintains the status quo. It is of interest as to why this replicative capacity and the ability to maintain total pancreatic cell mass cannot be channeled by the body to bring about regeneration of the pancreas without an external stimulus. We understand the phenomenon of organ homeostasis, where each tissue or organ has an inherent 'regulatory machinery' that regulates its own cell mass. The pancreas traces the damage in its milieu and responds by triggering limited islet neogenesis and replication [38]. However, this mechanism of cell mass maintenance is insufficient in cases of extreme damage, requiring external stimuli to initiate regeneration.
 |
|
Figure 2. Critical balance between accelerating and inhibitory factors affecting adult stem cell expansion and differentiation for triggering pancreatic regeneration. |
|
What are the external stimuli and when are they actually needed?
External stimuli are the inductive factors that trigger the cell machinery into replication/expansion of precursor cell populations and influence β-cell differentiation. In pancreatectomy, for instance, tissue injury induces a response to stimulate tissue healing and cellular regeneration. As is evident from current literature, the regeneration of any organ is a process where cells of varied lineages and undecided and decided cell fates together compensate for the lost cell mass. However, the existing sources of cell mass maintenance are inadequate to justify the regeneration of an entire organ in vivo. Hence it appears plausible to assume the presence of a yet unidentified stem cell population, which we would like to call naive precursor cells (NPC). These NPC may possess functional plasticity and replicative capacity to respond to appropriate stimulation, helping in regeneration to compensate for the lost β-cell mass. There are studies where the presence of similar types of cell populations has been illustrated, such as pancreatic multipotent progenitors [71] and small cells [72]. It is likely that these cells would undergo a steady change of state before adopting the fate of the mature β-cell population.
Taken together, these findings imply that the β-cell function may be restored endogenously and that the residual β-cell mass may be expanded in the diabetic host pancreas after the onset of disease. In recent years, cell-based therapy and regenerative medicine has been providing new therapeutic approaches for restoring organ functions lost due to trauma, disease or aging. To enhance tissue and organ regeneration, progenitor cells must be mobilized and provided with an appropriate niche to advance their development. To harness the power of regenerative medicine for diabetes therapy, more information is needed to define potential candidates and their niches. Investigating the cellular and molecular mechanisms regulating β-cell turnover and identifying potential therapeutic targets capable of enhancing human β-cell regeneration in vivo may help in translating research from bench to bedside. Such approaches will provide the necessary insights required to induce endogenous pancreatic regeneration.
Acknowledgments:
The authors wish to thank Dr. Anandwardhan Hardikar, Scientist 'D' NCCS for the critical comments and valuable inputs in the preparation of this manuscript.
References
- Bouckenooghe T, Vandewalle B, Moerman E, Danze PM, Lukowiak B, Muharram G, Kerr-Conte J, Gmyr V, Laine B, Pattou F. Expression of progenitor cell markers during expansion of sorted human pancreatic beta-cells. Gene Expr 2005. 12: 83-98. [DOD]
- Silver K, Tolea M, Wang J, Pollin TI, Yao F, Mitchell BD. The Exon1 Cys7Gly polymorphism within the Betcellulin gene is associated with Type2 Diabetes in African Americans. Diabetes 2005. 54:1179-1184. [DOD]
- Huotari MA, Palgi J, Otonkoski T. Growth factor-mediated proliferation and differentiation of insulin-producing INS-1 and RINm5F cells: identification of betacellulin as a novel beta-cell mitogen. Endocrinol 1998. 139:1494-1499. [DOD] [CrossRef]
- Lei L, Zhaohong Y, Mashaharu S, Kojima I. Activin A and Betacellulin: Effect on regeneration of pancreatic beta-cells in neonatal streptozotocin- treated rats. Diabetes 2004. 53:608-615. [DOD]
- Li L, Seno M, Yamada H, Kojima I. Betacellulin improves glucose metabolism by promoting conversion of intra-islet precursor cells to beta-cells in streptozotocin-treated mice. American Journal of Physiology Endocrinology and Metabolism 2003. 285:577-583. [DOD]
- Yoshikumi Y, Mashima H, Udea N, Ohno H, Suzuki J, Tanaka S, Hayashi M, Sekine N, Ohnishi H, Yasuda H, Iiri T, Omata M, Fujita T, Kojima I. Roles of CTPL/Sfxn3 family members in pancreatic islet. Journal of Cellular Biochemistry 2005. 95:1157-1168. [DOD] [CrossRef]
- Bolkent S, Yanardag R, Tabakoglu-Oguz A, Ozsoy-Sacan O. Effects of Chard extract (Beta vilgaris L. var. Cicla) extract on pancreatic beta-cells in streptozotocin–diabetic rats: a morphological and biochemical study. J Ethanopharmacol 2000. 73:251-259. [DOD] [CrossRef]
- Xiu LM, Miura AB, Yamamoto K, Kobayashi T, Song QH, Kitamura H, Cyong JC. Pancreatic islet regeneration by ephedrine in mice with streptozotocin-induced diabetes. Am J Chin Med 2001. 29:493-500. [DOD] [CrossRef]
- Vague P, Picq R, Bernal M, Lassmann-Vague V, Vialettes B. Effect of nicotinamide treatment on the residual insulin secretion in type 1 (insulin dependant) diabetic patients. Diabetologia 1989. 32:316-321. [DOD] [CrossRef]
- Fernandez-Alvarez J, Barbera A, Nadal B, Barcelo-Batllori S, Piquer S, Claret M, Guinovart JJ, Gomis R. Stable and functional regeneration of pancreatic beta-cell population in STZ-rats treated with tungstate. Diabetologia 2004. 47:470-477. [DOD] [CrossRef]
- Kim BM, Han YM, Shin YJ, Min BH, Park IS. Clusterin expression during regeneration of pancreatic islet cells in streptozotocin-induced diabetic rats. Diabetologia 2001. 44:2192-2202. [DOD] [CrossRef]
- Kato K, Isaji S, Kawarada Y, Hibasami H, Nakashima K. Effect of zinc administration on pancreatic regeneration after 80% pancreatectomy. Pancreas 1997. 14:158-165. [DOD]
- Taylor CG. Zinc, the pancreas, and diabetes: insight from rodent studies and future directions. Biomaterial 2005. 18:305-312. [DOD]
- Majumdar AP, Vesenka GD, Dubick MA. Acceleration of pancreatic regeneration by cholecytokinin in rats. Pancreas 1978. 2:199-204. [DOD]
- Morriset J. Intervention of GI neuropeptides in pancreatic growth and regeneration: a comparison with cholecytokinin. Journal of Physio. Pharmacol 2003. 54:127-141. [DOD]
- Fiorucci S, Bufalari A, Distrutti E, Lanfrancone L, Servoli A, Sarpi L, Federici B, Bartoli A, Morelli A, Moggi L. Bombesin-induced pancreatic regeneration in pigs is mediated by p46Shc/p52Shc and p42/p44 mitogen-activated protein kinase upregulation. Scand J Gastroenterol 1998. 33:1310-1320. [DOD] [CrossRef]
- Ledda-Columbano GM, Perra A, Pibiri M, Molotzu F, Columbano A. Induction of pancreatic acinar cell proliferation by thyroid hormone. Journal of Endocrinology 2005. 185:393-399. [DOD] [CrossRef]
- Brand SJ, Tagerud S, Lambert P, Magil SG, Tatarkiewicz K, Doiron K, Yan Y. Pharmacological treatment of chronic diabetes by stimulating pancreatic beta-cell regeneration with systemic co-administration of EGF and gastrin. Pharmacol Toxicol 2002. 91:414-420. [DOD] [CrossRef]
- Hayashi KY, Tamaki H, Handa K, Takahashi T, Kakita A, Yamashina S. Differentiation and proliferation of endocrine cells in the regenerating rat pancreas after 90% pancreatectomy. Arch Histol Cytol 2003. 66:163-174. [DOD] [CrossRef]
- Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999. 48:2270-2276. [DOD]
- De Leon DD, Deng S, Madani R, Ahima RS, Drucker DJ, Stoffers DA, Roleo F. Endogenous glucagon-like peptide-1 in islet regeneration after partial pancreatectomy. Diabetes 2003. 52:365-371. [DOD]
- Jetton TL, Liu YQ, Trotman WE, Nevin PW, Sun XJ, Leahy JL. Enhanced expression of insulin receptor substrate-2 and activation of protein kinase B/Akt in regenerating pancreatic duct epithelium of 60%-partial pancreatectomy rats. Diabetologia 2001. 44:2056-2065. [DOD] [CrossRef]
- Smith FE, Rosen KM, Villa-Komaroff L, Weir GC, Bonner-Weir S. Enhanced insulin-like growth actor-I gene expression in regenerating rat pancreas. Proc Natl Acad Sci USA 1991. 88:6152-6156. [DOD]
- Li L, Seno M, Yamada H, Kojima I. Promotion of beta-cell regeneration by betacellulin in 90% pancreatectomized rats. Endocrinology 2001. 142:5279-5385. [DOD]
- Yonemura Y, Takashima T, Miwa K, Miyazaki I, Yamamoto H, Okamoto H. Amelioration of diabetes mellitus in partially pancreatectomized rats by poly (ADP-ribose) synthase inhibitors. Evidence of islet beta-cell regeneration. Diabetes 1984. 33:401-404. [DOD]
- Sugiyama K, Yonemura Y, Okamoto H. Effects of poly (ADP-ribose) synthase inhibitors on B-cells of a canine pancreas after massive pancreatectomy. Pancreatol 1991. 8:85-95. [DOD]
- Gross DJ, Weiss L, Reibstein I, Van den Brand J, Okamoto H, Clark A, Slavin S. Amelioration of diabetes in nonobese diabetic mice with advanced disease by linomide-induced immunoregulation combined with reg protein treatment. Endocrinol 1998. 139:2369-2374. [DOD] [CrossRef]
- De Breuck S, Lardon J, Rooman I, Bouwens L. Netrin-expression in fetal and regenerating rat pancreas and its effect on the migration of human pancreatic duct and porcine islet precursor cells. Diabetologia 2003. 46:926-933. [DOD] [CrossRef]
- Rosenberg L. Induction of islet cell neogenesis in the adult pancreas: the partial duct obstruction model. Microsc Res Tech 1998. 43:337-346. [DOD]
- Hardikar AA, Karandikar MS, Bhonde RR. Effect of partial pancreatectomy on diabetic status in BALB/c mice. J Endocrinol 1999. 162:185-195. [DOD]
- Hardikar AA, Bhonde RR. Modulating experimental diabetes by treatment with cytosolic extract from the regenerating pancreas. Diabetes Res and Clin Pract 1999. 46:203-211. [DOD]
- Kanitkar M, Bhonde R. Existence of islet regenerating factors within the pancreas. Rev Diabetic Stud 2004. 1:185-192. [DOD]
- Swenne I, Andersson A. Effect of genetic background on the capacity for islet cell replication in mice. Diabetologia 1984. 27:464-467. [DOD] [CrossRef]
- Asanuma N, Kitamura T, Xu X, Sumitani S, Saito H, Kasayama S, Kouhara H, Kawase I. New analytical method for pancreas and liver regeneration: normalization of streptozotocin-induced hyperglycemia by retrograde injection of insulin producing cells. Endocr J 2002. 49:449-457. [DOD] [CrossRef]
- Yamamoto M, Yasuda M, Hori A, Arishima K, Eguchi Y. Recovery in the fetal pancreatic islet following fetal administration of streptozotocin in the rat in vivo and in vitro. Anat Rec A Discov Mol Cell Evol Biol 2004. 281:1319-1325. [DOD] [CrossRef]
- Guz Y, Nasir I, Teitelman G. Regeneration of pancreatic beta-cells from intra-islet precursor cells in an experimental model of diabetes. Endocrinology 2001. 142:4956-4968. [DOD] [CrossRef]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004. 429:41-46. [DOD] [CrossRef]
- Seaberg RM, Smukler SR, Kieffer TJ, Enikolopov G, Asghar Z, Wheeler MB, Korbutt G, van der Kooy D. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat Biotechnol 2004. 22:1115-1124. [DOD] [CrossRef]
- Bouwens L, Blay ED. Islet morphogenesis and stem cell markers in rat pancreas. J Histochem Cytochem 1996. 44:947-951. [DOD]
- Bouwens L, Lu WG, Krijger RD. Proliferation and differentiation in the human fetal endocrine pancreas. Diabetologia 1997. 40:398-404. [DOD] [CrossRef]
- Bertelli E, Regoli M, Orazioli D, Bendayan M. Association between islets of Langerhans and pancreatic ductal system in adult rat. Where endocrine and exocrine meet together? Diabetologia 2001. 44:575-584. [DOD]
- Bouwens L, Pipeleers DG. Extra-insular beta cells associated with ductules are frequent in adult human pancreas. Diabetologia 1998. 41:629-633. [DOD] [CrossRef]
- Dudek RW, Lawrence IE Jr, Hill RS, Johnson RC. Induction of islet cytodifferentiation by fetal mesenchyme in adult pancreatic ductal epithelium. Diabetes 1991. 40:1041-1048. [DOD]
- Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A 2000. 97:7999-8004. [DOD] [CrossRef]
- Elkin G, Prigozhina TB, Slavin S. Prevention of diabetes in nonobese diabetic mice by nonmyeloablative allogeneic bone marrow transplantation. Exp Hematol 2004. 32:579-584. [DOD] [CrossRef]
- Hess D, Li L, Martin M, Sakano S, Hill D, Strutt B, Thyssen S, Gray DA, Bhatia M. Bone marrow-derived stem cells initiate pancreatic regeneration. Nat Biotechnol 2003. 21:763-770. [DOD] [CrossRef]
- Ianus A, Holz GG, Theise ND, Hussain MA. In vivo derivation of glucose competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J Clin Invest 2003. 111:843-850. [DOD] [CrossRef]
- Scheffold YC, Weissman IL, Taylor C, Jerabek L, Burge MJ, Masek MA, Shizuru JA. Purified allogeneic hematopoietic stem cell transplantation blocks diabetes pathogenesis in NOD mice. Diabetes 2003. 52:59-68. [DOD]
- Lechner A, Yang YG, Blacken RA, Wang L, Nolan AL, Habener JF. No evidence for significant transdifferentiation of bone marrow into pancreatic beta-cells in vivo. Diabetes 2004. 53:616-623. [DOD]
- Choi JB, Uchino H, Azuma K, Iwashita N, Tanaka Y, Mochizuki H, Migita M, Shimada T, Kawamori R, Watada H. Little evidence of transdifferentiation of bone marrow-derived cells into pancreatic beta-cells. Diabetologia 2003. 46:1366-1374. [DOD] [CrossRef]
- Zorina TD, Subbotin VM, Bertera S, Alexander AM, Haluszczak C, Gambrell B, Bottino R, Styche AJ, Trucco M. Recovery of the endogenous beta-cell function in the NOD model of autoimmune diabetes. Stem Cells 2003. 21:377-388. [DOD] [CrossRef]
- Beilhack GK, Mathews V, Hanson PT, Ford E, Fujita J, Polonsky KS, Graubert TA. Recruitment of bone marrow-derived endothelial cells to sites of pancreatic beta-cell injury. Diabetes 2004. 53:91-98. [DOD]
- Banerjee M, Kumar A, Bhonde RR. Reversal of experimental diabetes by multiple bone marrow transplantation. Biochem Biophys Res Commun 2005. 328:318-325. [DOD] [CrossRef]
- Izumida Y, Aoki T, Yasuda D, Koizumi T, Suganuma C, Saito K, Murai N, Simizu Y, Hayashi K, Odaira M, Kusano T, Kushima M, Kusano M. Hepatocyte growth factor is constitutively produced by donor derived bone marrow cells and promotes regeneration of pancreatic beta-cells. Biochem Biophys Res Commun 2005. 333:273-282. [DOD] [CrossRef]
- Grompe M. Pancreatic-hepatic switches in vivo. Mech Dev 2003. 120:99-106. [DOD] [CrossRef]
- Zheng YW, Taniguchi H. Diversity of hepatic stem cells in the fetal and adult liver. Semin Liver Dis 2003. 23:337-348. [DOD] [CrossRef]
- Kojima H, Nakamura T, Fujita Y, Kishi A, Fujimiya M, Yamada S, Kudo M, Nishio Y, Maegawa H, Haneda M, et al. Combined expression of pancreatic duodenal box 1 and islet factor 1 induces immature enterocytes to produce insulin. Diabetes 2002. 51:1398-1408. [DOD]
- Fujita Y, Cheung AT, Kieffer TJ. Harnessing the gut to treat diabetes. Pediatr Diabetes 2004. 5(Suppl 2):57-69. [DOD] [CrossRef]
- Bjerknes M, Cheng H. Gastrointestinal Stem Cells. II. Intestinal stem cells. Am J Physiol Gastrointest Liver Physiol 2005. 289:381-387. [DOD] [CrossRef]
- Kodama S, Kuhtreiber W, Fujimura S, Dale EA, Faustman DL. Islet regeneration during the reversal of autoimmune diabetes in NOD mice. Science 2003. 302:1223-1227. [DOD] [CrossRef]
- Kodama S, Davis M, Faustman DL. Diabetes and stem cell researchers turn to the lowly spleen. Sci Aging Knowledge Environ 2005. 19:2. [DOD]
- Krapp A, Knofler M, Ledermann B, Burki K, Berney C, Zoerkler N, Hagenbuchle O, Wellauer PK. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev 1998. 12:3752-3763. [DOD]
- Ende N, Chen R, Reddi AS. Effect of human umbilical cord blood cells on glycemia and insulitis in type 1 diabetic mice. Biochem Biophys Res Commun 2004. 325:665-669. [DOD] [CrossRef]
- Ende N, Chen R, Reddi AS. Transplantation of human umbilical cord blood cells improves glycemia and glomerular hypertrophy in type 2 diabetic mice. Biochem Biophys Res Commun 2004. 321:168-171. [DOD] [CrossRef]
- Naruse K, Hamada Y, Nakashima E, Kato K, Mizubayashi R, Kamiya H, Yuzawa Y, Matsuo S, Murohara T, Matsubara T, Oiso Y, Nakamura J. Cord blood stem cells: Therapeutic neovascularization using cord blood-derived endothelial progenitor cells for diabetic neuropathy. Diabetes 2005. 54:1823-1828. [DOD]
- Wei JP, Zhang TS, Kawa S, Aizawa T, Ota M, Akaike T, Kato K, Konishi I, Nikaido T. Human amnion-isolated cells normalize blood glucose in streptozotocin-induced diabetic mice. Cell Transplant 2003. 12:545-552. [DOD]
- Luo X, Yang H, Kim IS, Saint-Hilaire F, Thomas DA, De BP, Ozkaynak E, Muthukumar T, Hancock WW, Crystal RG, Suthanthiran M. Systemic transforming growth factor-beta1 gene therapy induces Foxp3+ regulatory cells, restores self-tolerance, and facilitates regeneration of beta-cell function in overtly diabetic nonobese diabetic mice. Transplantation 2005. 79:1091-1096. [DOD] [CrossRef]
- Zhang YQ, Cleary MM, Si Y, Liu G, Eto Y, Kritzik M, Dabernat S, Kayali AG, Sarvetnick N. Inhibition of activin signaling induces pancreatic epithelial cell expansion and diminishes terminal differentiation of pancreatic beta-cells. Diabetes 2004. 53:2024-2033. [DOD]
- Tian C, Bagley J, Cretin N, Seth N, Wucherpfennig KW, Iacomini J. Prevention of type 1 diabetes by gene therapy. J Clin Invest 2004. 114:969-978. [DOD] [CrossRef]
- Morral N. Gene therapy for type 1 diabetes. New approaches. Minerva Med 2004. 95:93-104. [DOD]
- Suzuki A, Nakauchi H, Taniguchi H. Prospective isolation of multipotent pancreatic progenitors using flow-cytometric cell sorting. Diabetes 2004. 53:2143-2152. [DOD]
- Petropavlovskaia M, Rosenberg L. Identification and characterization of small cells in the adult pancreas: potential progenitor cells? Cell Tissue Res 2002. 310:51-58. [DOD]
This article has been cited by other articles:

|
A review on endogenous regenerative technology in periodontal regenerative medicine
Chen FM, Zhang J, Zhang M, An Y, Chen F, Wu ZF
Biomaterials 2010. 31(31):7892-7927
|
|

|
Human umbilical cord blood as an emerging stem cell therapy for diabetes mellitus
Reddi AS, Kuppasani K, Ende N
Curr Stem Cell Res Ther 2010. 5(4):356-361
|
|

|
Antihyperglycemic activity of catharanthus roseus leaf powder in streptozotocin-induced diabetic rats
Rasineni K, Bellamkonda R, Singareddy SR, Desireddy S
Pharmacogn Res 2010. 2(3):195-201
|
|

|
Islet neogenesis-associated protein pentadecapeptide (INGAP-PP): mechanisms involved in its effect upon beta-cell mass and function
Madrid V, Del Zotto H, Maiztegui B, Raschia MA, Alzugaray ME, Boschero AC, Barbosa HC, Flores LE, Borelli MI, Gagliardino JJ
Regul Pept 2009. 157(1-3):25-31
|
|

|
Can splenocytes enhance pancreatic beta-cell function and mass in 90% pancreatectomized rats fed a high fat diet?
Park S, Hong SM, Ahn IS
Life Sci 2009. 84(11-12):358-363
|
|

|
The influence of pancreas-derived stem cells on scaffold based skin regeneration
Salem H, Ciba P, Rapoport DH, Egana JT, Reithmayer K, Kadry M, Machens HG, Kruse C
Biomaterials 2009. 30(5):789-796
|
|
|
Surgical conditions of the Canine and Feline pancreas
Havlicek M, Franklin A
Aust Vet Pract 2008. 38(4):146-152
|
|

|
Recent progress on normal and malignant pancreatic stem/progenitor cell research: therapeutic implications for the treatment of type 1 or 2 diabetes mellitus and aggressive pancreatic cancer
Mimeault M, Batra SK
Gut 2008. 57(10):1456-1468
|
|

|
Nicotinamide: A cytoprotectant against streptozotocininduced diabetic damage in wistar rat brains
Ibrahim SS, Rizk SM
Afr J Biochem Res 2008. 2(8):174-180
|
|

|
Stem cell potential for type 1 diabetes therapy
Roche E, Ramirez M, Ramirez-Castillejo C, Gomez-Mauricio G, Uson J
Cent Europ J Biol 2007. 2(4):449-480
|
|

|
Advancements of stem-cell therapy for type 1 diabetes mellitus
Li H, We J, Mu C
Chin Bull Life Sci 2007. 19(4):401-408
|
|
|


)
)

