Diabetic Perspectives
| Rev Diabet Stud,
2006,
3(1):39-46 |
DOI 10.1900/RDS.2006.3.39 |
Apoptosis in Autoimmune Diabetes: The Fate of β-Cells in the Cleft between Life and Death
Charles Sia1, Arno Hänninen2
1United Biomedical Inc., 25 Davids Drive, Hauppauge, New York 11788, USA.
2Department of Medical Microbiology, University of Turku, Kiinamyllynkatu 13, 20520 Turku, Finland.
Address correspondence to: Charles Sia, e-mail: csia@unitedbiomedical.com.
Keywords: type 1 diabetes, beta-cell, apoptosis, Fas
Abstract
Cytokine-induced β-cell death is the end-stage event in the pathogenesis of autoimmune diabetes. Beside cytokines, several pro-apoptotic pathways mediated through nitric oxide, reactive oxygen species, glucose and Fas ligation can be involved, suggesting that programmed cell death (PCD) is a critical aspect in this process. The apoptotic program is activated by the utilization of the Fas/Fas-ligand (FasL) axis in the interrelation of T and β-cells. Evidence for this mechanism arose from the finding that β-cells in NOD mice could be protected from apoptosis by blocking the Fas-FasL pathway. Glucose is a regulator of Fas expression on human β-cells and elevated glucose levels may contribute to accelerated β-cell destruction by constitutively expressed FasL independently of the autoimmune reaction. It can thus be concluded that immunological, as well as metabolic, pathways may act in concert to cause β-cell destruction. Much experimental work has been carried out to manipulate β-cells in transgenic mice expressing apoptosis modulators in islets. For example, the transcription factor, nuclear factor-κB (NF-κB), promotes the expression of several β-cell genes, including pro- and anti-apoptotic genes. The prevention of cytokine-induced gene expression of several NF-κB targets, such as inducible nitric oxide synthase, Fas, and manganese superoxide dismutase can prevent β-cell death. Thus, modulating the expression of apoptotic mediators may significantly affect the end-stage outcome of autoimmune diabetes and could thus be a potential avenue for clinical therapy, even though currently existing findings remain exploratory due to the restrictions of transgenic mouse models.
Introduction
Tissue homeostasis maintains the body in a stable equilibrium, involving the interplay of cellular growth, differentiation and death. The process is regulated by programmed cell death (PCD), which is switched on by the activation of an intrinsic suicide program to remove superfluous, aging, infected and mutated cells from the body’s tissue [1, 2]. PCD, also called apoptosis, refers to a set of events within a cell which leads to the breakdown of chromosomal DNA and the cessation of metabolic activity. It can be induced by different stimuli such as deprivation of growth and survival factors, i.e. death by neglect, or by ligation of surface receptors that contain a death domain, then called receptor-mediated apoptosis [3]. Apoptosis is an active process mediated by serine proteases called caspases, so-called due to their specificity for amino acid motifs containing an aspartic acid cleaved by these enzymes. Caspases exist within a cell as zymogens, i.e. inactive precursors called procaspases. Similarly to the blood clotting system or the complement system, activation of a few molecules of an upstream procaspase can activate a growing number of effector caspases situated downstream in the activation cascade. Full activation of this cascade leads to nuclear condensation accompanied by DNA fragmentation [4, 5]. Shrinkage of the whole cell then occurs, with parts of the cell disintegrating into smaller apoptotic fragments. Macrophages finally clear these fragments and the residual cell bodies by phagocytosis [6].
Apoptotic processes remove unwanted cells without releasing their cellular contents into the extracellular space. These processes have an important function in avoiding unnecessary immune responses, because the existence of, for example double-stranded DNA, uric acid and lysosomal proteases, may be recognized as foreign implicating the generation of inflammatory responses. A failure in the clearance of cellular contents during apoptosis is likely to play a role in the pathogenesis of systemic autoimmune diseases, such as systemic lupus erythematosus (SLE) and other rheumatic connective tissue diseases [7]. Failures in caspase activation, in turn, lead to certain forms of the autoimmune lymphoproliferative syndrome (ALPS), which is characterized by massive accumulation of activated T cells in peripheral lymphoid tissues and a generalized inflammatory and wasting condition [8, 9]. Thus, failures in the clearance of apoptotic β-cells could play a critical role in the generation of unintentional immune responses towards islets, and a failure of the immune system to control islet-reactive T cells could involve defective activation of apoptosis during the process of β-cell destruction. Therefore, the pathways mediating and controlling apoptotic cell death are of obvious relevance in the pathogenesis of T1DM.
The role of death domain-containing receptors and their ligands in T1DM
Death domain receptors and T cell apoptosis
Tumor necrosis factor (TNF) is a pleiotropic cytokine which can induce activation, differentiation, proliferation or death of cells of the immune system. Screening of the expressed sequence tag (EST) database almost a decade ago gave rise to the discovery of a whole family of structurally related cytokines and their receptors that were thus named the TNF and TNFR (TNF receptor) superfamilies [10]. Some members of the TNFR superfamily contain a so-called death-domain (DD) in their cytoplasmic tail, and depending on the TNFR they are associated with, they have been named TRADD (TNF-receptor associated death domain) and FADD (Fas-associated death domain)/FLICE [11, 12]. DDs are able to recruit procaspase 8 (an upstream caspase) upon ligand binding to the receptor to form the death-inducing signaling complex (DISC) and thus activate receptor-mediated PCD within the cell (Figure 1) [4]. Receptor-mediated PCD is important in the downscaling of an immune response after clonal expansion and activation of antigen-specific T cells. This process is called activation-induced cell death (AICD) [13]. In general, T cells are resistant to receptor-mediated PCD until they are fully activated. This resistance is due to intracellular proteins that inhibit the formation of death-signaling complexes within the cell. Their expression is downregulated later during T cell activation when T cells themselves also start to express the ligand for Fas (FasL, CD95L) [14, 15].
In human subjects of recent onset T1DM, FasL is expressed on CD8+ and CD4+ T cells and on macrophages that have infiltrated into their islet tissues [16]. It is conceivable that only a thin line separates two very different fates of activated T cells, i.e. survival and the ability to exert an effector function, and the fate of becoming apoptotic. The factors regulating this subtle balance are of obvious importance in the regulation of T cell-mediated immune responses, and may also play an important role in regulating the actions of islet-infiltrating T cells. Although a lot of these T cells are likely to become apoptotic in islets, they seem to be able to orchestrate inflammation and cytotoxicity at a level sufficient to induce β-cell death before PCD takes place. It is also possible that the inflammatory environment in infiltrated islets supports their survival through cytokines, e.g. of the TNF family, and cell-cell contacts exerting anti-apoptotic signals, e.g. through TNFR that do not contain death domains.
Death domain receptors and β-cell apoptosis
Proinflammatory cytokines, such as IL-1α and IFN-γ, upregulate expression of Fas on islet cells upon in vitro exposure (Figure 1) [17, 18]. Fas is not expressed on islet cells of normal subjects, but its expression on islet β-cells has been detected by histological examination on pancreatic biopsy specimens of patients with recent-onset T1DM [17, 19]. Enhanced Fas expression may be due to the release of the inducible form of nitric oxide synthase (iNOS) by infiltrated macrophages and autoreative CD4+ and CD8+ T cells [19, 20]. While Fas-ligand (FasL, CD95L) has even been reported to be expressed by cultured islet cells of normal subjects [17, 19, 21], it is conceivable that ligation of Fas by FasL expressed by individual and neighboring β-cells could be one mechanism driving β-cells to apoptosis (fratricide-suicide). Unlike FasL, Fas is not expressed on islets of normal subjects, but its expression is upregulated in vitro upon exposure to proinflammatory cytokines, such as IL-1α and IFN-γ (Figure 1) [17, 19, 22]. The combination of these cytokines produced by pancreas-infiltrated macrophages and autoreative CD4+ and CD8+ T cells may induce β-cells to release iNOS, which possibly leads to enhanced Fas expression and drive them to die by apoptosis [23-25].
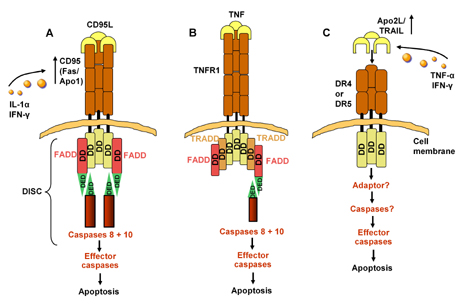 |
 |
Figure 1. Apoptotic pathways. A. Apoptosis signaling by CD95. DD: death domain. DED: death effector domain. CD95L (FasL) is a homotrimeric molecule. Each CD95L trimer binds three CD95 molecules. CD95 and CD95L play an important role in different types of physiologic apoptosis: 1. peripheral deletion of activated mature T cells; 2. killing of targets such as virus-infected cells or, in the case of T1DM, β-cells cells by cytotoxic T cells; 3. killing of inflammatory cells. The adapter protein FADD binds through its own death domain to the clustered receptor death domains. FADD contains a "death effector domain" that binds to an analogous domain repeated in tandem within the zymogen form of caspase 8 (also called FLICE). Upon recruitment by FADD, caspases 8 and 10 then activates downstream effector caspases committing the cell to apoptosis [4]. IL-1α and IFN-γ upregulate expression of Fas on islet cells [17]. B. Proapoptotic and antiapoptotic signaling by TNFR1. Unlike CD95L, TNF triggers apoptosis unless protein synthesis is blocked, which suggests the preexistence of cellular factors that can suppress the apoptotic stimulus generated by TNF. TNF trimerizes TNFR1 upon binding, inducing association of the receptors' death domains. Subsequently, an adapter termed TRADD binds through its own death domain to the clustered receptor death domains. C. Signaling by DR4 and DR5 and its modulation by decoy receptors. Overexpression of DR4 or DR5, which bind Apo2L, triggers apoptosis. There are divergent results regarding the required adaptor for the death domain of DR4 and DR5 [4]. Exposure to TNF-α and INF-γ leads to upregulation of TRAIL that binds to DR4 on β-cells and subsequently leads to a higher rate of β-cell apoptosis [27]. Figure modified according to [4]. |
|
Expression of TNF-related apoptosis-inducing ligand (TRAIL), which interacts with the death receptor DR4 [26], is upregulated in apoptosis-sensitive, insulin-secreting MIN6 islet β-cells upon exposure to TNF-α and IFN-γ in vitro (Figure 1) [27, 28]. TRAIL is able to induce stronger cytotoxicity and a higher rate of apoptosis in different β-cell lines than FasL and TNF-α. β-cell antigen-specific CD4+ and CD8+ clones express TRAIL, suggesting that these cells are potential sources of TRAIL-inducing β-cell destruction [29]. NOD mice at the stage of overt diabetes also overexpress TRAIL in their pancreatic islets as compared to younger prediabetic mice [28]. There is still sparse evidence regarding a clear correlation between its expression and β-cell apoptosis in diabetes but several findings point to this hypothesis. In a recent clinical study human pancreases of children with acute established and recent-onset T1DM exhibited the presence of TRAIL, while none could be observed in normal islets [30].
Fas-FasL interaction mediates apoptosis of β-cells during T1DM development
Studies conducted in NOD mice suggest that T1DM development in these animals is a 2-stage process [31]. The first phase is asymptomatic, involving a loss of immunological tolerance which favors the infiltration of islet-specific lymphoid cells including CD4+, CD8+ and B cells, as well as macrophages and dendritic cells into the pancreatic islets. The development of insulitis, including the destruction of β-cells by apoptosis, occurs in the second stage. The T cell-mediated cytotoxic destruction of β-cells takes place through two distinctive mechanisms, one is perforin-based, the other Fas-based. In this event, Fas is upregulated on β-cells by cytokine release and Fas-positive β-cells interact with FasL-bearing CD4 T cells. On the other hand, cytolytic killing of β-cells by CD8 effector cells is provoked through the perforin/gran-zyme B pathway [32]. Proinflammatory cytokines released by islet-infiltrated lymphocytes drive β-cells to produce higher levels of iNOS which, in turn, enhance their Fas expression [22]. However, cytokine-induced β-cell apoptosis also happens in a NO-independent manner [33, 34]. Therefore, perpetual β-cell apoptosis occurs predominantly in a Fas-dependent manner engaged by FasL-bearing autoreactive T cells.
Additional results regarding the role of Fas-FasL-mediated apotosis in T1DM showed that Fas is expressed at a low quantity on β-cells in 125-day-old prediabetic animals but not in islet tissues of 35-day-old young NOD mice [35]. These observations suggest that Fas-FasL interaction is a minor contributor to β-cell destruction in the initial stage of the disease. On the other hand, we may interpret these results in the way that Fas expression on β-cells within the early stage of diabetes is too low to become detected by islet-infiltrating autoreactive cells to a sufficient degree as to provoke extensive islet cell destruction. Only after cytokine-induced upregulation of Fas expression does the apoptotic Fas-FasL-pathway become commanding. Increased production of islet antigens released from damaged β-cells may further costimulate the proliferation of autoreactive T cells to exert a more aggressive diabetogenicity. In support of this notion is the finding that in vitro incubation of NOD mouse islets with IL-1α and IFN-γ leads to a more intensive Fas expression on β-cells which allows them to interact better with FasL-bearing autoreactive T cells, thus rendering them to undergo apoptosis [36].
The finding that FasL was detected on β-cells of neither NOD nor Fas-deficient MRL-lpr/lpr islets, while both mouse strains were equally susceptible to cytokine-induced β-cell death, implies that an alternative mechanism other than Fas-FasL interaction may be important in diabetes development [37]. Perforin-positive offspring derived from perforin-deficient C57BL/6 mice backcrossed to NOD mice succumb to the development of overt diabetes more rapidly than their perforin-negative littermates. Furthermore, perforin-negative animals exhibited a greater resistance to developing cyclophosphamide-induced diabetes [38]. In lymphocytic choriomeningitis virus (LCMV)-glycoprotein (GP) transgenic mice, which express virus-GP on islet β-cells, LCMV infections led to the generation of GP-specific T cells and induced diabetes development, but perforin-deficiency has been found to confer resistance to diabetes development in this model despite Fas expression [39]. We can thus conclude from the existing findings that the perforin pathway must be a second critical apoptotic pathway in the development of autoimmune diabetes.
However, there are also contradictory results regarding the perforin apoptotic pathway. In 8.3-TCR transgenic mice, β-cells were destroyed by diabetogenic T cells via the Fas-FasL system, while perforin knock-out has been observed to be insufficient in protecting the animals from developing diabetes [40]. Fas-deficient NOD.lpr/lpr mice have been found to be resistant to T cell-mediated apoptosis and remain free from developing spontaneous diabetes despite expressing perforin (Figure 2) [37]. However, protection in these animals may be attributed to the existence of abnormal FasL-expressing cells in the host capable of mediating apoptosis against the transferred autoreactive T cells rather than to a reduced or lacking Fas-FasL interaction [41]. In a different study, a population of suppressor cells bearing the CD4-CD8-B220+ phenotype has been found in the NOD.lpr/lpr mice, suggesting that B220+ suppressor cells contribute to a down-regulation of diabetogenic T cells in Fas-deficient NOD.lpr/lpr mice [42].
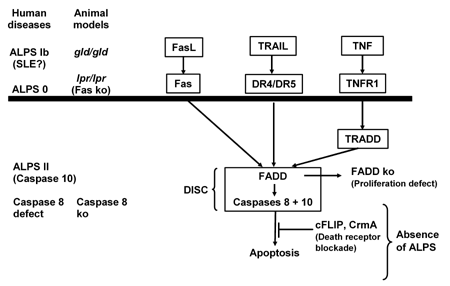 |
|
Figure 2. Human diseases and animal models of death ligand/receptor signaling defects. ALPS Ib is the human genetic counterpart of the gld mice and corresponds to FasL blockade in mice. NOD mice heterozygous for the FasL mutation gld express low FasL levels on their T lymphocytes and exhibit delayed progression to diabetes. Fas-deficient NOD.lpr/lpr mice are resistant to T cell-mediated apoptosis. Transgenic animals expressing an inhibitor of caspase 8 do not develop lymphoproliferation although all death-receptor pathways are blocked. Upregulation of cFLIP has the same effect and secures insulin secretion in the insulin-secreting cell line βTc-Tet. Transgenic expression of CrmA in RIP-NOD mice inhibits caspases 8 and 10 and leads to delayed diabetes onset induced by diabetogenic spleen cells. Figure modified according to [9]. |
|
Apart from the abnormal FasL expression in NOD.lpr/lpr mice that exerts an anti-diabetic activity against diabetogenic T cells, we can conclude that the majority of studies carried out in rodent diabetes models showed that both FasL expression and the perforin pathway correlate with diabetes development. NOD mice with β-cells overexpressing FasL are less resistant to diabetogenic T cells and are found to develop diabetes at an accelerated rate compared to the wild type NOD counterparts [42]. Furthermore, higher FasL expression is detected in pancreatic islet tissues of cyclophosphamide-induced diabetic mice and female NOD mice as they progress to develop insulitis and become diabetic [43, 44]. NOD mice heterozygous for the FasL mutation gld resulting in the expression of suboptimal levels of functional FasL on their T lymphocytes develop insulitis but exhibit delayed progression to diabetes (Figure 2) [45].
Death signal modulation for T1DM intervention
The understanding that Fas-FasL-mediated apoptosis induced by autoreactive T cells against islet β-cells leads to diabetic insulitis has prompted several studies to address the question as to whether disruption of this pathway may protect from diabetes development in rodent disease models.
Intervention in the interaction of effector and target cells
Blocking Fas-FasL interaction by anti-FasL antibodies is effective in preventing NOD mice at 2-4 weeks but not at 5-15 weeks of age from becoming diabetic. Adoptive cell transfer experiments revealed that inhibiting FasL on diabetogenic CD4 but not CD8 T cells is critical for mediating protection since administration of anti-FasL antibodies is effective in preventing diabetes development induced by diabetogenic CD4+ and not CD8+ T cell transfer [46].
A recombinant fusion protein FAS-Fc consisting of murine Fas and the Fc moiety of human IgG1 administrated in multiple doses has been found to protect NOD mice from developing cyclophosphamide-induced diabetes. Unexpectedly, instead of FasL blockade interfering with Fas-FasL interaction, the treatment was found to preserve the functional integrity of regulatory T cells that contributed to the resistance of diabetes development in the animals [47].
Restoring self tolerance
The active form of vitamin D, 1α,25-dihydroxy-vitamin D3 (1α,25(OH)2D3), has been reported to mediate immunoregulatory functions and as such it was used as an immunosuppressive reagent in transplantation [48]. NOD mice treated with the compound gained resistance to insulitis and subsequent diabetes development [49]. Protection in these cases can be attributed to a mechanism that facilitates an improved positive selection of thymocytes and the induction of tolerogenic dendritic cells contributing to the downregulation of autoreactive T cell activity [50].
Functional modulation of autoreactive T cells
Costimulation via CD28/B7 between T cells and antigen-presenting cells is a necessary consecutive pathway for the activation of T cells [51]. Recently, a new member of the TNF superfamily, TNFSF14 which is also called LIGHT, expressed on activated T cells that binds the herpes virus entry mediator (HVEM), has been shown to promote T cell proliferation via its interaction with LIGHT [52, 53]. This mechanism is also associated with the proliferation of autoreactive T cells in the development of autoimmune diseases such as T1DM. Blockade of LIGHT by treating 5- to 6-week-old female NOD mice with HVEM-Ig (100 μg per mouse) for a short term (a duration 3 weeks) led to a significant prevention of disease development [54].
Targeted expression of FADD-like IL-1β-converting enzyme (FLICE) to prevent β-cell apoptosis
Insulin secretion of murine and rat islet cells is affected by TNF-α in a concentration-dependent manner [55, 56]. However, a combination of TNF-α with IL-1α and IFN-γ is needed to induce apoptosis of mouse islet cells [57]. TNF-α-induced apoptosis can be inhibited by a variety of mediators, including FLICE-inhibitory proteins [58]. In vitro studies with the insulin-secreting cell line βTc-Tet showed that upregulated expression of cellular FLIP (cFLIP) led to the blockade of caspase 8 activity that rendered the cells resistant to apoptosis mediated by TNF-α, IL-1-α and IFN-γ (Figure 2). This strategy also prevented a significant reduction in insulin secretion induced by exposure of the cells to TNF-α alone [59]. These results encourage further investigation as to whether overexpression of cFLIP in human islet cells may improve the acceptance of islet implants.
Targeted expression of cytokine response modifier A (CrmA) to prevent β-cell apoptosis
CrmA is a cowpox virus-encoded protein that belongs to the serine protease inhibitor family. CrmA is able to inhibit caspases 8 and 10 to prevent a variety of mammalian cell types, including pancreatic islet cells, from undergoing apoptosis (Figure 2) [60]. Transgenic expression of CrmA in RIP-NOD mice (rat insulin promoter driven adenovirus E9 transgenic NOD mice) is unable to prevent streptozotocin-induced spontaneous diabetes development, but is effective in preventing diabetes development induced by diabetogenic spleen cells [61]. In a recent study, targeted expression of CrmA to pancreatic β-cells of NOD mice at birth led to a substantial delay and reduction of diabetes onset after adoptive transfer of diabetic spleen cells or diabetogenic CD8 T-cell clones (Figure 2) [62]. In the light of these findings, modulation of CrmA expression does not appear to be a feasible approach for preventing T1DM development, but overexpression of CrmA in islet β-cells could be tested as an additional procedure for prolonging the life of islet grafts.
Immunomodulation with TRAIL to prevent β-cell death
Pancreatic islet β-cells of NOD mice at the stage of overt insulitis show upregulated TRAIL expression. However, β-cells of these animals do not undergo apoptosis upon exposure to TRAIL, suggesting that enhanced TRAIL expression is unlikely to contribute to β-cell apoptosis. On the other hand, TRAIL is able to render human autoantigen-specific T cells hyporesponsive without driving them to undergo apoptosis [63]. In another study that involves the administration of a recombinant soluble TRAIL receptor, sDR5, into cyclophosphamide-treated NOD mice and NOD.Scid mice adoptively transferred with diabetogenic T cells, the animals are found to develop diabetes at a significantly accelerated rate than untreated mice [28]. This has been attributed to the blocking of the anti-diabetogenic property of TRAIL with sDR5 rather than to enhanced β-cell apoptosis.
Conclusion
In summary, apoptosis mediated by the engagement of death receptors with their respective ligands contributes critically to T1DM development. New experimental systems address the question as to whether modulating the expression of these molecules may lead to the design of future therapeutic regimens for the treatment of T1DM. It is still too early to relate the effectiveness of any of these approaches or a combination of them to the therapeutic potential of T1DM. However, with increasing understanding in the mechanisms provoking β-cell death and survival modulation of the life-span of activated, autoreactive T cells through their death receptors can open promising avenues for the development of new T1DM intervention strategies.
Acknowledgments:
This work was developed with support of the Society for Biomedical Diabetes Research (SBDR, 2006).
References
- Meier P, Finch A, Evan G. Apoptosis in development. Nature 2000. 407:796-801. [DOD] [CrossRef]
- Lincz LF. Deciphering the apoptotic pathway: all roads lead to death. Immunol Cell Biol 1998. 76(1):1-19. [DOD] [CrossRef]
- Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem 1998. 254(3):439-459. [DOD] [CrossRef]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998. 281:1303-1306. [DOD] [CrossRef]
- Lang F, Szabo I, Lepple-Wienhues A, Siemen D, Gulbins E. Physiology of Receptor-Mediated Lymphocyte Apoptosis. News Physiol Sci 1999. 14:194-200. [DOD]
- Savill J, Fadok V. Corpse clearance defines the meaning of death. Nature 2000. 407:784-788. [DOD] [CrossRef]
- Roberts KM, Rosen A, Casciola-Rosen LA. Methods for inducing apoptosis. Methods Mol Med 2004. 102:115-128. [DOD]
- Martin DA, Zheng L, Siegel RM, Huang B, Fisher GH, Wang J, Jackson CE, Puck JM, Dale J, Straus SE, Peter ME, Krammer PH, Fesik S, Lenardo MJ. Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci U S A 1999. 96(8):4552-4557. [DOD] [CrossRef]
- Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways. Cell Death Differ 2003. 10(1):124-133. [DOD] [CrossRef]
- Tribouley C, Wallroth M, Chan V, Paliard X, Fang E, Lamson G, Pot D, Escobedo J, Williams LT. Characterization of a new member of the TNF family expressed on antigen presenting cells. Biol Chem 1999. 380(12):1443-1447. [DOD] [CrossRef]
- Baker SJ, Reddy EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene 1996. 12(1):1-9. [DOD]
- Darnay BG, Aggarwal BB. Early events in TNF signaling: a story of associations and dissociations. J Leukoc Biol 1997. 61(5):559-566. [DOD]
- Fas SC, Fritzsching B, Suri-Payer E, Krammer PH. Death receptor signaling and its function in the immune system. Curr Dir Autoimmun 2006. 9:1-17. [DOD]
- Oberg HH, Lengl-Janssen B, Kabelitz D, Janssen O. Activation-induced T cell death: resistance or susceptibility correlate with cell surface fas ligand expression and T helper phenotype. Cell Immunol 1997. 181(1):93-100. [DOD] [CrossRef]
- Marsters SA, Sheridan JP, Pitti RM, Brush J, Goddard A, Ashkenazi A. Identification of a ligand for the death-domain-containing receptor Apo3. Curr Biol 1998. 8:525-528. [DOD] [CrossRef]
- Suarez-Pinzon WL, Power RF, Rabinovitch A. Fas ligand-mediated mechanisms are involved in autoimmune destruction of islet beta cells in non-obese diabetic mice. Diabetologia 2000. 43(9):1149-1156. [DOD] [CrossRef]
- Stassi G, Todaro M, Richiusa P, Giordano M, Mattina A, Sbriglia S, Lo Monte A, Buscemi G, Galluzzo A, Giordano C. Expression of apoptosis-inducing CD95 (Fas/Apo-1) on human beta-cells sorted by flow-cytometry and cultured in vitro. Transplant Proc 1995. 27:3271-3275. [DOD]
- Darville MI, Liu D, Chen MC, Eizirik DL. Molecular regulation of Fas expression in beta-cells. Diabetes 2001. 50(Suppl 1):S83. [DOD]
- Moriwaki M, Itoh N, Miyagawa J, Yamamoto K, Imagawa A, Yamagata K, Iwahashi H, Nakajima H, Namba M, Nagata S, Hanafusa T, Matsuzawa Y. Fas and Fas ligand expression in inflamed islets in pancreas sections of patients with recent-onset Type I diabetes mellitus. Diabetologia 1999. 42(11):1332-1340. [DOD] [CrossRef]
- Rabinovitch A. An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Diabetes Metab Rev 1998. 14(2):129-151. [DOD] [CrossRef]
- Hanke J. Apoptosis in cultured rat islets of langerhans and occurrence of Bcl-2, Bak, Bax, Fas and Fas ligand. Cells Tissues Organs 2001. 169(2):113-124. [DOD] [CrossRef]
- Stassi G, De Maria R, Trucco G, Rudert W, Testi R, Galluzzo A, Giordano C, Trucco M. Nitric oxide primes pancreatic beta cells for Fas-mediated destruction in insulin-dependent diabetes mellitus. J Exp Med 1997. 186(8):1193-1200. [DOD] [CrossRef]
- McDaniel ML, Corbett JA, Kwon G, Hill JR. A role for nitric oxide and other inflammatory mediators in cytokine-induced pancreatic beta-cell dysfunction and destruction. Adv Exp Med Biol 1997. 426:313-319. [DOD]
- Delaney CA, Pavlovic D, Hoorens A, Pipeleers DG, Eizirik DL. Cytokines induce deoxyribonucleic acid strand breaks and apoptosis in human pancreatic islet cells. Endocrinology 1997. 138(6):2610-2614. [DOD] [CrossRef]
- Loweth AC, Williams GT, James RF, Scarpello JH, Morgan NG. Human islets of Langerhans express Fas ligand and undergo apoptosis in response to interleukin-1beta and Fas ligation. Diabetes 1998. 47(5):727-732. [DOD]
- Pan G, O'Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science 1997. 276(5309):111-113. [DOD] [CrossRef]
- Ishizuka N, Yagui K, Tokuyama Y, Yamada K, Suzuki Y, Miyazaki J, Hashimoto N, Makino H, Saito Y, Kanatsuka A. Tumor necrosis factor alpha signaling pathway and apoptosis in pancreatic beta cells. Metabolism 1999. 48(12):1485-1492. [DOD] [CrossRef]
- Mi QS, Ly D, Lamhamedi-Cherradi SE, Salojin KV, Zhou L, Grattan M, Meagher C, Zucker P, Chen YH, Nagle J, Taub D, Delovitch TL. Blockade of tumor necrosis factor-related apoptosis-inducing ligand exacerbates type 1 diabetes in NOD mice. Diabetes 2003. 52(8):1967-1975. [DOD]
- Ou D, Metzger DL, Wang X, Huang J, Pozzilli P, Tingle AJ. TNF-related apoptosis-inducing ligand death pathway-mediated human beta-cell destruction. Diabetologia 2002. 45(12):1678-1688. [DOD] [CrossRef]
- Cheung SS, Metzger DL, Wang X, Huang J, Tai J, Tingle AJ, Ou D. Tumor necrosis factor-related apoptosis-inducing ligand and CD56 expression in patients with type 1 diabetes mellitus. Pancreas 2005. 30(2):105-114. [DOD] [CrossRef]
- Andre-Schmutz I, Hindelang C, Benoist C, Mathis D. Cellular and molecular changes accompanying the progression from insulitis to diabetes. Eur J Immunol 1999. 29(1):245-255. [DOD] [CrossRef]
- Kägi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science 1994. 265:528-530. [DOD]
- Liu D, Pavlovic D, Chen MC, Flodstrom M, Sandler S, Eizirik DL. Cytokines induce apoptosis in beta-cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS-/-). Diabetes 2000. 49(7):1116-1122. [DOD]
- Zumsteg U, Frigerio S, Hollander GA. Nitric oxide production and Fas surface expression mediate two independent pathways of cytokine-induced murine beta-cell damage. Diabetes 2000. 49(1):39-47. [DOD]
- Thomas HE, Darwiche R, Corbett JA, Kay TW. Evidence that beta cell death in the nonobese diabetic mouse is Fas independent. J Immunol 1999. 163:1562-1569. [DOD]
- Mandrup-Poulsen T, Bendtzen K, Nerup J, Dinarello CA, Svenson M, Nielsen JH. Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia 1986. 29:63-67. [DOD]
- Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, Makino S, Nagata S, Kono N, Matsuzawa Y. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med 1997. 186(4):613-618. [DOD] [CrossRef]
- Kägi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice. J Exp Med 1997. 186(7):989-997. [DOD] [CrossRef]
- Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of the anti-self (virus) immune response. Cell 1991. 65:319-331. [DOD] [CrossRef]
- Amrani A, Verdaguer J, Anderson B, Utsugi T, Bou S, Santamaria P. Perforin-independent beta-cell destruction by diabetogenic CD8(+) T lymphocytes in transgenic nonobese diabetic mice. J Clin Invest 1999. 103(8):1201-1209. [DOD]
- Kim S, Kim KA, Hwang DY, Lee TH, Kayagaki N, Yagita H, Lee MS. Inhibition of autoimmune diabetes by Fas ligand: The paradox is solved. J Immunol 2000. 164:2931-2936. [DOD]
- Hamad AR, Mohamood AS, Trujillo CJ, Huang CT, Yuan E, Schneck JP. B220+ double negative-T cells suppress polyclonal T cell activation by a Fas-independent mechanism that involves inhibition of IL-2 production. J Immunol 2003. 171:2421-2426. [DOD]
- Reddy S, Ross JM. Fas and Fas ligand immunoexpression in pancreatic islets of NOD mice during spontaneous and cyclophosphamide-accelerated diabetes. Ann NY Acad Sci 2003. 1005:166-169. [DOD] [CrossRef]
- Cao JY, Wang H. Role of Fas-FasL in insulitis in nonobese diabetic mouse. Chin Med J (Engl) 2004. 117(4):615-617. [DOD]
- Suarez-Pinzon WL, Power RF, Rabinovitch A. Fas ligand-mediated mechanisms are involved in autoimmune destruction of islet beta cells in non-obese diabetic mice. Diabetologia 2000. 43:1149-1156. [DOD] [CrossRef]
- Nakayama M, Nagata M, Yasuda H, Arisawa K, Kotani R, Yamada K, Chowdhury SA, Chakrabarty S, Jin ZZ, Yagita H, Yokono K, Kasuga M. Fas/Fas ligand interactions play an essential role in the initiation of murine autoimmune diabetes. Diabetes 2002. 51:1391-1397. [DOD]
- Mahiou J, Walter U, Lepault F, Godeau F, Bach JF, Chatenoud L. In vivo blockade of the Fas-Fas ligand pathway inhibits cyclophosphamide-induced diabetes in NOD mice. J Autoimmunity 2001. 16(4):431-440. [DOD] [CrossRef]
- Becker BN, Hullett DA, O'Herrin JK, Malin G, Sollinger HW, DeLuca H. Vitamin D as immunomodulatory therapy for kidney transplantation. Transplantation 2002. 74(8):1204-1206. [DOD] [CrossRef]
- Decallonne B, Mathieu C. Defective activation-induced cell death in NOD T lymphocytes: 1,25-dihydroxyvitamin D3 restores defect. Ann N Y Acad Sci 2003. 1005:176-177. [DOD] [CrossRef]
- Decallonne B, van Etten E, Overbergh L, Valckx D, Bouillon R, Mathieu C. 1alpha,25-dihydroxyvitamin D3 restores thymocyte apoptosis sensitivity in non-obese diabetic (NOD) mice through dendritic cells. J Autoimmun 2005. 24(4):281-289. [DOD] [CrossRef]
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996. 14:233-258. [DOD] [CrossRef]
- Kwon BS, Tan KB, Ni J, Oh KO, Lee ZH, Kim KK, Kim YJ, Wang S, Gentz R, Yu GL, Harrop J, Lyn SD, Silverman C, Porter TG, Truneh A, Young PR. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J Biol Chem 1997. 272(22):14272-14276. [DOD] [CrossRef]
- Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 1998. 8(1):21-30. [DOD] [CrossRef]
- Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, Tamada K, Chen L, Fu YX. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest 2001. 108(12):1771-1780. [DOD] [CrossRef]
- Campbell IL, Iscaro A, Harrison LC. IFN-gamma and tumor necrosis factor-alpha: cytotoxicity to murine islets of Langerhans. J Immunol 1988. 141:2325-2329. [DOD]
- Thomas HE, Ming L, Grell M, Darwiche R, Volodin L, Kay TW. Tumor necrosis factor-alpha-activated cell death pathways in NIT-1 insulinoma cells and primary pancreatic beta cells. Endocrinology 1999. 140:3219-3227. [DOD] [CrossRef]
- Dunger A, Cunningham JM, Delaney CA, Lowe JE, Green MH, Bone AJ, Green IC. Tumor necrosis factor-alpha and interferon-gamma inhibit insulin secretion and cause DNA damage in unweaned-rat islets: extent of nitric oxide involvement. Diabetes 1996. 45:183-189. [DOD]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature 1997. 388:190-195. [DOD] [CrossRef]
- Cottet S, Dupraz P, Hamburger F, Dolci W, Jaquet M, Thorens B. cFLIP protein prevents tumor necrosis factor-alpha-mediated induction of caspase-8-dependent apoptosis in insulin-secreting betaTc-Tet cells. Diabetes 2002. 51(6):1805-1814. [DOD]
- Dbaibo GS, Hannun YA. Cytokine response modifier A (CrmA): a strategically deployed viral weapon. Clin Immunol Immunopathol 1998. 86(2):134-140. [DOD] [CrossRef]
- Hugues S, Mougneau E, Ferlin W, Jeske D, Hofman P, Homann D, Beaudoin L, Schrike C, Von Herrath M, Lehuen A, Glaichenhaus N. Tolerance to islet antigens and prevention from diabetes induced by limited apoptosis of pancreatic beta cells. Immunity 2002. 16(2):169-181. [DOD] [CrossRef]
- Millet I, Wong FS, Gurr W, Wen L, Zawalich W, Green EA, Flavell RA, Sherwin RS. Targeted expression of the anti-apoptotic gene CrmA to NOD pancreatic islets protects from autoimmune diabetes. J Autoimmun 2006. 26(1):7-15. [DOD] [CrossRef]
- Lunemann JD, Waiczies S, Ehrlich S, Wendling U, Seeger B, Kamradt T, Zipp F. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J Immunol 2002. 168:4881-4888. [DOD]
This article has been cited by other articles:

|
Mechanisms of toxic effect of streptozotocin on beta-cells in the islets of langerhans
Pisarev VB, Snigur GL, Spasov AA, Samokhina MP, Bulanov AE
Bull Exp Biol Med 2009. 148(6):937-939
|
|

|
Prevention and management of hyperglycemia after pancreas transplantation
Egidi MF, Lin A, Bratton CF, Baliga PK
Curr Opin Organ Transplant 2008. 13(1):72-78
|
|

|
Expression and distribution of S-100 protein, CD83 and apoptosis-related proteins (Fas, FasL and Bcl-2) in thyroid tissues of autoimmune thyroid diseases
Xu WC, Chen SR, Huang JX, Zheng ZC, Chen LX, Lin JJ, Li YG
Eur J Histochem 2007. 51(4):291-300
|
|

|
Stem cells: a potential cure for type I and II diabetes
Myers L
DUJS 2007. 9(2)
|
|
|


)
)

