Diabetic Perspectives
| Rev Diabet Stud,
2006,
3(2):88-95 |
DOI 10.1900/RDS.2006.3.88 |
Death Pathways in T Cell Homeostasis and Their Role in Autoimmune Diabetes
Matthew A. Gronski1,  2 2
1Beirne Carter Center for Immunology Research, University of Virginia, Lane Road, Charlottesville, VA 22908, USA.
2SBDR, Gneisenaustr. 86, 47057 Duisburg, Germany.
Address correspondence to: Matthew Gronski, e-mail: gronski@virginia.edu
Keywords: type 1 diabetes, T cell, apoptosis, programmed cell death, mitochondria, death receptor, fas
Abstract
T cell apoptosis is a process necessary for central and peripheral tolerance. It ensures the proper removal of autoreactive T cells during thymic development as well as T cell homeostasis and the downregulation of immune responses against antigens in the periphery. Thus it is essential for the prevention of autoimmunity. Apoptotic pathways can be triggered by intrinsic (mitochondria-based) and extrinsic (receptor-based) stimuli. Both pathways involve a cascade of proteolytic enzymes called caspases whose activation commits the cell to death. In the periphery, autoreactive lymphocytes can be silenced by developmental arrest (anergy), or deleted by programmed cell death (apoptosis) through receptor-based activation-induced cell death (AICD). Central tolerance seems to rely more heavily on the mitochondria-based, T cell receptor (TCR)-stimulated apoptotic pathway, since thymocytes lacking the pro-apoptotic Bcl-2 family member Bim are resistant to TCR-induced apoptosis. Furthermore, defects in the intrinsic pathway of apoptosis may impair clonal deletion of autoreactive T cells. Several animal models exist in which impaired apoptosis results in autoimmunity. Here, we discuss data that suggest defects in T cell apoptosis in type 1 diabetes mellitus.
Introduction
T cell homeostasis and fully functional T cell compartments are critical to achieve an effective defense against foreign antigens and to avoid overreactivity against self-antigens [1]. This is achieved through processes mediating proliferation and elimination of lymphocytes. Signaling through antigen receptors promotes survival, proliferative expansion and differentiation of useful and deletion of useless and dangerous T cells [2].
Apoptosis is necessary to control the development of T cells and to maintain the T cell repertoire in a balanced state (homeostasis). It thus acts in two ways. Firstly, T cell development and maturation in the thymus require education and selection mechanisms. T cells that can recognize and remain unresponsive against self major histocompatibility complex (MHC) and self-antigens are positively and those that cannot recognize or that are even activated by interactions with self-MHC and self-antigens are negatively selected and die by apoptosis [3]. Thymic apoptosis programs are thus necessary for the selection of functional, correctly equipped and sensitive T cells, while at the same time, to avoid the escape of degenerated (e.g. insensitive or equipped with wrong markers), incompletely developed (e.g. with missing pre-T cell receptor (TCR) α or β) or autoreactive T cells [4, 5].
Secondly, during immune responses, activated and pathogen-specific peripheral T cells, which matured in the thymus and were exported to the periphery, undergo extensive expansion and proliferation and develop effector functions, such as cytotoxicity or cytokine production [6]. When infectious or foreign tissue cells are eliminated by a T cell-mediated immune response, most of the activated T cells are removed by a process called programmed cell death (PCD), also called apoptosis, in order to prevent damage of own tissue by these effector cells and the released cytotoxic molecules [7, 8]. The shift towards a pro-apoptotic milieu at the end of an immune response is achieved by limiting the provision of growth factors such as IL-2, and a decrease in the expression of anti-apoptotic Bcl-2 members, a mitochondria-dependent protein family containing members that can be pro- or anti-apoptotic [9, 10]. The remaining T cells survive. Upon withdrawal of stimulation, they assume a memory phenotype and live on as trained quiescent lymphocytes in peripheral lymph nodes [11]. For the survival of mature resting T cells and those with memory functions, costimulation by CD28 ligation and IL-2 expression, which is increased by costimulation, together with anti-apoptotic Bcl-2 expression is necessary [12-14]. In combination with other mechanisms, including developmental arrest or induction of unresponsiveness (anergy), PCD of autoreactive lymphocytes safeguards immunological tolerance to self-antigens and assists in the development and maintenance of an effective and tolerant immune system [2].
In autoimmune diabetes, activated CD4+ and CD8+ T cells proceed to kill self-tissue after an early event of pancreatic inflammation by peripheral blood monocytes and macrophages [15]. Even though it is not clear whether autoimmunity is initiated as a consequence of an environmental trigger such as a virus infection, molecular mimicry, abnormal TCR or something different, genetic predisposition is suspected to play a decisive role in the development of type 1 diabetes (T1DM) [16, 17]. This phenomenon points to an intrinsic failure in antigen recognition, processing or T cell selection and elimination. In any case, islet antigens, such as glutamic acid decarboxylase (GAD), insulin, proinsulin and tyrosine phosphatase (IA-2), are recognized as foreign and are antagonized by a T-cell-mediated immune response [18, 19]. After activation of islet-specific CD4+ and CD8+ T cell populations by cell-to-cell contact with APC and their subsequent proliferation, the destruction of pancreatic β-cells occurs. The process involves a large number of self-reactive T cells and the question arises why these cells escape thymic or peripheral tolerance mechanisms. It seems that in diabetic patients either thymic selection or PCD after an immune response is defective. Defects in thymic selection could be observed in NOD mice [20] and there is also evidence for defects in peripheral activation-induced cell death (AICD) after immune response [21, 22]. Beside PCD, other mechanisms mediating tolerance are also suspected to bear defects in T1DM, e.g. disturbances in the activity of regulatory T cells of the CD4+CD25+ phenotype, which control potentially self-reactive T cells [23]. However, this article focuses on the mechanisms that control PCD and discusses possible failures in this process that could play a role in T1DM development as well as potential strategies to overcome them.
Regulation of T cell homeostasis by apoptosis and the role of death signals
Naïve and memory T cells exist as silent lymphocytes in the periphery where they are maintained by the provision of stimulation, growth factors such as IL-2 and a high internal level of anti-apoptotic Bcl-2 [14, 24, 25]. Following an immune response most of the activated T cells need to be deleted by apoptosis, which is induced by a switch from an apoptosis-resistant to an apoptosis-sensitive state. This can be mediated by cytokines, death receptors and pro-apoptotic proteins [26].
The process of PCD can be initiated by two different but interlinked pathways: an intrinsic, mitochondria-dependent and an extrinsic, receptor-induced pathway. Despite different modes of initiation, an activation of a cascade of proteolytic enzymes, termed cysteine proteases or simply caspases, occurs. Once initiator caspases (8, 9 and 10) are activated, the cell is committed to death. Both pathways play critical roles in terminating immune responses [27].
Death receptor-induced apoptosis
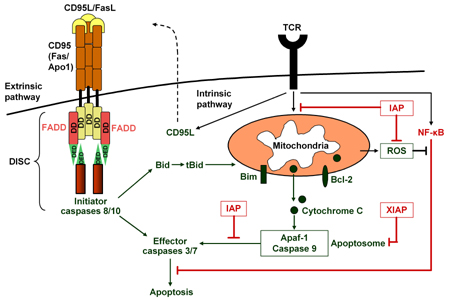
The extrinsic pathway of apoptosis, also called activation-induced cell death (AICD), is initiated by ligation of membrane-bound trimeric death receptors such as CD95/Fas, tumor necrosis factor receptor (TNFR) and death receptors (DR) 4 and 5, which are expressed upon TCR stimulation [28, 29]. Death receptor ligation either by CD95L (FasL), TNF or TNFR-related apoptosis-inducing ligand (TRAIL, also called Apo2L) leads to caspase 8 activation, procaspase 3 cleavage and subsequently to cell death [26, 29]. Caspases exist within a cell as zymogens, i.e. inactive precursors called procaspases. Activation of a procaspase can activate effector caspases (3 and 7) downstream in the activation cascade. Full activation of these effector caspases eventually leads to nuclear condensation, DNA fragmentation and cell death (Figure 1) [30, 31].
 |
 |
Figure 1. The extrinsic and intrinsic pathways of apoptosis. The extrinsic pathway is initiated by ligation of membrane-bound trimeric death receptors such as CD95/Fas. Adaptor molecules activate initator caspases through the DISC. The intrinsic pathway is induced by TCR stimulation and mitochondira membrane permeabilization. |
|
Death receptors contain a so-called death domain (DD) in their cytoplasmic tail and, depending on the receptor the DDs are associated with; they have been named Fas-associated death domain (FADD) and TNFR-associated death domain (TRADD). DDs are able to recruit initiator caspases 8 and 10 upon binding through their death effector domain (DED) to an analogous domain repeated in tandem within the zymogen form of the caspases. Sensitivity to FasL- as well as TNF-mediated cell death is thus controlled by the so-called death-inducing signaling complex (DISC), which contains the adaptor protein FADD and the initiator caspases 8 and 10. TNF-mediated apoptosis includes the TRADD in addition to the DISC (Figure 1) [26, 30].
AICD is important to downscale immune responses after activation and clonal expansion of antigen-specific T cells [32]. T cells are resistant to receptor-mediated apoptosis until they are fully activated. This resistance is due to intracellular proteins that inhibit or block death signaling within the cell. The caspase 8-homologous FLICE (Fas-associated death-domain-like interleukin 1β-converting enzyme) can be blocked by cellular FLICE inhibitory protein (c-FLIP) [33-35]. If death receptor-induced stimulation is blocked or not sufficient to execute full effector caspase activity, caspase 8 can mediate the cleavage of the pro-apoptotic Bcl-2 member Bid towards truncated Bid (tBid) and thereby initiate the intrinsic pathway of apoptosis [36, 37].
Mitochondria-dependent apoptosis
The mitochondria-de-pendent apoptotic pathway is mainly controlled by the balance between pro- and anti-apoptotic members of the Bcl-2 protein family [38]. A critical event in the apoptotic process is the change of the mitochondrial membrane potential provoked by the Bcl-2 family of proteins that either protect against or induce membrane depolarization and permeabilization [39]. Depolarization, once induced, is a reversible but strong initiator of the intrinsic apoptotic program. Subsequently, cytochrome C is released by the mitochondria and causes the adaptor protein apoptotic protease-activating factor 1 (Apaf-1) to activate caspase 9 [40]. The activated complex containing cytochrome C, Apaf-1 and caspase 9 is called the apoptosome and initiates cell death. In several cell types, the pro-apoptotic Bcl-2 family member Bim on mitochondrial membranes is involved in TCR-induced apoptosis. Bim is upregulated by p38 and JNK and causes mitochondrial membrane depolarization and thereby initiates the intrinsic death pathway (Figure 1) [41]. Bim is a critical initiator of mitochondria-dependent, stress-induced cell death and T cell homeostasis and serves as a barrier against autoimmune disease. Bim-deficiency severely impairs the apoptosis of autoreactive thymocytes [42].
Pro-apoptotic pathways are antagonized by anti-apoptotic vigors to ensure that apoptosis only occurs in response to appropriate death signals. So-called inhibitors of apoptosis (IAP) belong to this class of intrinsic proteins and a prominent member is the X-linked IAP (XIAP). IAP is able to set a threshold for the induction of apoptosis and inhibits effector caspases from becoming activated (Figure 1) [43, 44].
Apoptosis is also controlled by the transcription factor nuclear factor κB (NF-κB) pathway. During T cell development, activation of NF-κB provides a strong signal for cell survival and proliferation in response to TCR stimulation [45]. NF-κB is a negative regulator of PCD also in response to TNF-α and similar cytokines [46]. On the other hand, inhibition of NF-κB has a pro-apoptotic effect, particularly increasing the susceptibility to TNF-α-induced death by the increase of reactive oxygen species (ROS) [47, 48]. Increased ROS suppress Bcl-2 expression and results in cell death via the intrinsic apoptosis pathway [49].
T cell deletion in the thymus
T cell deletion works at two main stages: in the thymus during central tolerance and in the periphery following an immune response. Differentiation of thymocytes is characterized by the cell surface expression of proteins, such as CD4 and CD8. Bone marrow progenitors that enter the thymus initially do not express CD4 or CD8 and are referred to as double negative (DN) CD4-CD8- thymocytes. Following T cell receptor (TCR) β chain rearrangement, only thymocytes expressing a functionally rearranged TCRβ chain are selected to continue maturation and upregulate both CD4 and CD8. At this CD4+CD8+-double positive (DP) stage, TCRα chain rearrangement is initiated and functionally rearranged TCRα chains are expressed on the cell surface together with the TCRβ chain to obtain a functional TCRαβ for recognizing antigens. Interactions between the TCR expressed by DP thymocytes and self-MHC molecules mediate survival or death. Thymocytes expressing TCR that do not interact with self-MHC molecules die within a few days. Thymocytes expressing TCRs that are able to interact with intermediate affinity with self-MHC molecules are rescued from death by a process called positive selection. Positive selection ensures that only T cells that are able to recognize peptides presented on self-MHC molecules are exported to the periphery. Thymocytes expressing TCRs recognizing class I MHC molecules downregulate CD4 and become cytotoxic CD8+ single positive (SP) cells, while CD4+ helper T cells arise as a result of interactions between TCR and class II MHC molecules. A strong interaction between TCR and self-peptide/MHC leads to the elimination of thymocytes by a process called negative selection [50, 51].
What happens to autoreactive thymocytes when they receive a strong signal through the TCR? Clonal deletion has been shown to be one of the main mechanisms that eliminates autoreactive cells [52-54]. Further evidence for clonal deletion as a mechanism of negative selection was provided by TCR transgenic mice [55, 56]. Together, these studies clearly demonstrate that thymocytes expressing self-reactive TCRs are clonally eliminated during development.
A potential problem with central tolerance is the limited expression of peripheral/tissue specific antigens in the thymus. It has become clear, however, that certain cells of the thymic stroma express peripheral tissue specific antigens that promote the elimination of self-reactive T cells [57, 58]. Studies have suggested that a putative transcription factor, the autoimmune regulator (AIRE) protein, is expressed in rare specialized cells called medullary epithelial cells (MEC). A mutation in this protein in human patients may lead to the development of the multiorgan autoimmune endocrine disease APECED [59, 60]. Since both AIRE and peripheral antigens are expressed in MEC, it was speculated and later confirmed by microarray technology, that AIRE controls the expression of these antigens [61]. To study the role of AIRE, AIRE-deficient mice were generated by two independent groups [61, 62]. These mice developed lymphocytic infiltrates and autoantibodies directed against a number of peripheral organs and tissues, such as the salivary gland, retina, pancreas, ovary, stomach and thyroid. Therefore, central tolerance includes mechanisms to induce tolerance of T cells specific for tissue antigens expressed outside the thymus.
T cell deletion to yield peripheral tolerance
Because not all autoreactive thymocytes are deleted during development, peripheral tolerance mechanisms exist to limit autoimmunity. Deletion of autoreactive T cells is one of these mechanisms. The mechanism of tolerance of mature T cells by clonal elimination was demonstrated in vivo using different approaches [63-65]. In the first report, injection of cells expressing the superantigen Mls-1a into thymectomized Mls-1b mice resulted in the expansion of Mls-1a reactive Vβ6+CD4+ T cells, followed by deletion of these cells [63]. Further demonstrations of clonal deletion as a mechanism of peripheral tolerance to self-antigens was provided using transgenic mouse models [66-68].
The importance of deletion as a mechanism of peripheral tolerance has also been suggested in mice deficient for genes involved in apoptosis such as Fas and FasL. The natural mutant lpr mouse (lymphoproliferative), which carries a mutation in the TNF-family receptor Fas and the FasL mutant gld mouse (generalized lymphoproliferative disorder), develop lymphadenopathy, splenomegaly and spontaneous autoimmunity [69, 70]. A similar phenotype and autoimmunity is seen in autoimmune lymphoproliferative syndrome (ALPS) patients (also known as Canale Smith Syndrome), most of whom have a mutation in Fas [71-73]. However, the role of Fas in peripheral clonal deletion and autoimmunity remains controversial. While some studies have pointed to a role for Fas/FasL in AICD of mature T cells but not thymocytes [74, 75], other studies found Fas to be dispensable for peripheral deletion [76-80]. Numerous other examples of molecules involved in apoptosis exist whose deregulation can result in autoimmunity [81-83].
Defects in T cell apoptosis in T1DM
Similarly, defects in apoptosis have also been suggested in diabetes. Studies using NOD mice have shown that normal thymic negative selection in these mice is impaired. It has been suggested that in NOD thymocytes the FLICE-inhibitory protein is upregulated upon TCR engagement which may result in the prevention of normal T cell deletion [20]. A further study using the BDC2.5 transgenic T cells on a NOD background suggests that a number of other apoptotic genes are differentially upregulated in NOD thymocytes during negative selection. Interestingly, on the NOD background, a different set of genes was up- or downregulated, rather than the same genes to different degrees. For example, the anti-apoptotic gene bcl2 was increased more than 2-fold on the NOD background compared to controls [84]. The authors then located Idd loci and genes responsible for the negative selection defect. It turned out that, besides bcl2, PD1 (another protein involved in apoptosis) was also implicated in the control of immune tolerance. Although very interesting, neither of the above-mentioned studies proved a causal relationship that impaired negative selection is the trigger or cause of diabetes in NOD mice.
Some studies have also suggested defects in peripheral AICD that follows immune responses [85, 86]. Lower levels and/or activity of caspase 8, as well as Fas/FasL were suggested as reasons for this difference. Furthermore, c-FLIP expression persists in NOD T cells. In general, there is a limited amount of evidence for abnormal T cell apoptosis in the literature. It is possible that the resistance to AICD arises from the defective proliferative response of NOD T cells, which can be overcome by IL-4 [22]. Similarly, the low level of FasL in NOD mice does not correlate with studies carried out in humans where no linkage of polymorphisms in the fasl gene to diabetes were found [87].
Overcoming defects in T cell apoptosis in order to reverse T1DM
How could potentially impaired T cell apoptosis processes in diabetes be reversed? This may be achieved either directly or indirectly. For example, injection of non-specific agents such as BCG or complete Freund’s adjuvant (CFA) prevents diabetes in NOD mice [88]. In an experiment carried out by Qin and coworkers, BCG immunization induced the production of TNF-α, IFN-γ and IL-4 by splenocytes, while, at the same time, it increased the expression of Fas, FasL and TNFR on T cells resulting in their apoptosis. The same effect could be reproduced by the systemic administration of the Th1 cytokines in these mice [88]. Problematically, such systemic treatments reduce the number of T cells of all specificities, not just the autoimmune ones, and may potentially leave the organism more susceptible to side effects such as infections or tumors.
A more specific or direct approach may involve increasing the concentration of negatively selecting peptides in the thymus. Peptides directly involved in the activation of autoreactive T cells (insulin, GAD, IA-2, ICA512) may be expressed at higher concentrations in both the thymus and the periphery. However, at this time, the technology available to achieve this effectively is limited and/or yet to be shown as being safe. Furthermore, this strategy can only be considered in subjects susceptible to develop diabetes and not in patients who have already developed the disease.
In cases of specific overabundance of anti-apoptotic factors (e.g. Bcl-2 in NOD mice, as discussed above), molecules could be developed that would target and block the function of these anti-apoptotic molecules. This is currently being tested in cancer patients [89]. However, the side effects may be still too great to justify the use of such compounds in diabetes patients. Hence, highly specific inhibitors need to be developed. An example of a more physiologic compound is vitamin D which has also been shown to work in NOD mice in preventing diabetes [90]. Although its mechanism of action is unclear, vitamin D enhances the susceptibility of thymocytes to apoptosis. Whether this approach would also be applicable in humans remains to be determined.
Although interesting and promising, many of the abovementioned treatments may be difficult in humans, as currently, there is no clear way of determining who will develop diabetes in the future and for whom such immunomodulatory techniques would be useful until clinical disease develops. However, because clinical diabetes is believed to occur when more than 90% of β-cells have been destroyed, it leaves a small window to counteract and potentially reverse the autoimmune process. These techniques may also be useful in the future in cases of autologous islet transplants from, for example, stem cell sources where the immune system would still be potentially autoreactive towards β-cells and tolerizing regimens would be necessary to prevent a recurrence of diabetes.
Conclusion
Apoptosis is crucial for the maintenance of tolerance and the prevention of autoimmunity. However, as of today, there is limited evidence to suggest that faulty apoptotic mechanisms are responsible for the development of diabetes. Although the evidence for a negative selection defect in the thymus is stronger than for one in the periphery, diabetes appears to be a more complicated disease involving a multitude of factors. Some of these may be components of the apoptotic machinery and would thus be interesting to investigate in the future. Thus, further studies will have to delineate apoptotic defects and to associate them with diabetes development, before potential treatment approaches can be considered.
References
- Marleau AM, Sarvetnick N. T cell homeostasis in tolerance and immunity. J Leukoc Biol 2005. 78(3):575-584. [DOD] [CrossRef]
- Strasser A, Bouillet P. The control of apoptosis in lymphocyte selection. Immunol Rev 2003. 193:82-92. [DOD] [CrossRef]
- Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis - immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol 1999. 17:221-253. [DOD] [CrossRef]
- Sebzda E, Mariathasan S, Ohteki T, Jones R, Bachmann MF, Ohashi PS. Selection of the T cell repertoire. Annu Rev Immunol 1999. 17:829-874. [DOD] [CrossRef]
- Paroli M, Barnaba V. Mechanisms of CD8+ T cell peripheral tolerance to our own antigens. Front Biosci 2005. 10:1628-1634. [DOD]
- Brown IE, Mashayekhi M, Markiewicz M, Alegre ML, Gajewski TF. Peripheral survival of naive CD8+ T cells. Apoptosis 2005. 10(1):5-11. [DOD] [CrossRef]
- Pinkoski MJ, Green DR. Apoptosis in the regulation of immune responses. J Rheumatol Suppl 2005. 74:19-25. [DOD]
- Konig R, Shen X, Maroto R, Denning TL. The role of CD4 in regulating homeostasis of T helper cells. Immunol Res 2002. 25(2):115-130. [DOD] [CrossRef]
- Broome HE, Dargan CM, Krajewski S, Reed JC. Expression of Bcl-2, Bcl-x, and Bax after T cell activation and IL-2 withdrawal. J Immunol 1995. 155(5):2311-2317. [DOD]
- Kreisel D, Sankaran D, Wells AD, Turka LA. Interleukin-2-mediated survival and proliferative signals are uncoupled in T lymphocytes that fail to divide after activation. Am J Transplant 2002. 2(2):120-128. [DOD] [CrossRef]
- Harbertson J, Biederman E, Bennett KE, Kondrack RM, Bradley LM. Withdrawal of stimulation may initiate the transition of effector to memory CD4 cells. J Immunol 2002. 168(3):1095-1102. [DOD]
- Benczik M, Gaffen SL. The interleukin (IL)-2 family cytokines: survival and proliferation signaling pathways in T lymphocytes. Immunol Invest 2004. 33(2):109-142. [DOD] [CrossRef]
- Collette Y, Razanajaona D, Ghiotto M, Olive D. CD28 can promote T cell survival through a phosphatidylinositol 3-kinase-independent mechanism. Eur J Immunol 1997. 77:3283-3289. [DOD]
- Ahmed R, Gray D. Immunological memory and protective immunity: Understanding their relation. Science 1996. 77:54-60. [DOD] [CrossRef]
- Andre I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad Sci U S A 1996. 93(6):2260-2263. [DOD] [CrossRef]
- Todd JA, Reed PW, Prins JB, Bain SC, Palmer SM, Cordell HJ, Pritchard LE, Ghosh S, Cornall RJ, Aitman TJ, et al. Dissection of the pathophysiology of type 1 diabetes by genetic analysis. Autoimmunity 1993. 15(Suppl):16-17. [DOD]
- Sia C, Weinem M. Genetic susceptibility to type 1 diabetes in the intracellular pathway of antigen processing - a subject review and cross-study comparison. Rev Diabetic Stud 2005. 2(1):40-52. [DOD] [CrossRef]
- Yoon JW, Jun HS. Cellular and molecular pathogenic mechanisms of insulin-dependent diabetes mellitus. Ann N Y Acad Sci 2001. 928:200-211. [DOD]
- Sarugeri E, Dozio N, Meschi F, Pastore MR, Bonifacio E. T cell responses to type 1 diabetes related peptides sharing homologous regions. J Mol Med 2001. 79(4):213-220. [DOD] [CrossRef]
- Kishimoto H, Sprent J. A defect in central tolerance in NOD mice. Nat Immunol 2001. 2(11):1025-1031. [DOD] [CrossRef]
- Decallonne B, Mathieu C. Defective activation-induced cell death in NOD T lymphocytes: 1,25-dihydroxyvitamin D3 restores defect. Ann N Y Acad Sci 2003. 1005:176-177. [DOD] [CrossRef]
- Aoki CA, Borchers AT, Ridgway WM, Keen CL, Ansari AA, Gershwin ME. NOD mice and autoimmunity. Autoimmun Rev 2005. 4(6):373-379. [DOD] [CrossRef]
- Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, Ten S, Sanz M, Exley M, Wilson B, Porcelli S, Maclaren N. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest 2002. 109(1):131-140. [DOD] [CrossRef]
- Benczik M, Gaffen SL. The interleukin (IL)-2 family cytokines: survival and proliferation signaling pathways in T lymphocytes. Immunol Invest 2004. 33(2):109-142. [DOD] [CrossRef]
- Collette Y, Razanajaona D, Ghiotto M, Olive D. CD28 can promote T cell survival through a phosphatidylinositol 3-kinase-independent mechanism. Eur J Immunol 1997. 77:3283-3289. [DOD]
- Arnold R, Brenner D, Becker M, Frey CR, Krammer PH. How T lymphocytes switch between life and death. Eur J Immunol 2006. 36(7):1654-1658. [DOD] [CrossRef]
- Green DR. Overview: apoptotic signaling pathways in the immune system. Immunol Rev 2003. 193:5-9. [DOD] [CrossRef]
- Krueger A, Fas SC, Baumann S, Krammer PH. The role of CD95 in the regulation of peripheral T-cell apoptosis. Immunol Rev 2003. 193:58-69. [DOD] [CrossRef]
- Janssen O, Sanzenbacher R, Kabelitz D. Regulation of activation-induced cell death of mature T-lymphocyte populations. Cell Tissue Res 2000. 301(1):85-99. [DOD] [CrossRef]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998. 281(5381):1305-1308. [DOD] [CrossRef]
- Lang F, Szabo I, Lepple-Wienhues A, Siemen D, Gulbins E. Physiology of receptor-mediated lymphocyte apoptosis. News Physiol Sci 1999. 14:194-200. [DOD]
- Konig R, Shen X, Maroto R, Denning TL. The role of CD4 in regulating homeostasis of T helper cells. Immunol Res 2002. 25(2):115-130. [DOD] [CrossRef]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature 1997. 388(6638):190-195. [DOD] [CrossRef]
- Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem 2001. 276(23):20633-20640. [DOD] [CrossRef]
- Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J Biol Chem 1999. 274(32):22532-22538. [DOD] [CrossRef]
- Strasser A, Puthalakath H, Bouillet P, Huang DC, O'Connor L, O'Reilly LA, Cullen L, Cory S, Adams JM. The role of bim, a proapoptotic BH3-only member of the Bcl-2 family in cell-death control. Ann N Y Acad Sci 2000. 917:541-548. [DOD]
- Esposti MD. The roles of Bid. Apoptosis 2002. 7(5):433-440. [DOD] [CrossRef]
- Grimaldi M, Denizot M, Espert L, Robert-Hebmann V, Biard-Piechaczyk M. Mitochondria-dependent apoptosis in T-cell homeostasis. Curr Opin Investig Drugs 2005. 6(11):1095-1102. [DOD]
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998. 1366(1-2):177-196. [DOD] [CrossRef]
- Le Bras M, Rouy I, Brenner C. The modulation of inter-organelle cross-talk to control apoptosis. Med Chem 2006. 2(1):1-12. [DOD] [CrossRef]
- Harada H, Grant S. Apoptosis regulators. Rev Clin Exp Hematol 2003. 7(2):117-138. [DOD]
- Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 2002. 415(6874):922-926. [DOD] [CrossRef]
- de Graaf AO, de Witte T, Jansen JH. Inhibitor of apoptosis proteins: new therapeutic targets in hematological cancer? Leukemia 2004. 18(11):1751-1759. [DOD]
- Conte D, Liston P, Wong JW, Wright KE, Korneluk RG. Thymocyte-targeted overexpression of xiap transgene disrupts T lymphoid apoptosis and maturation. Proc Natl Acad Sci U S A 2001. 98(9):5049-5054. [DOD] [CrossRef]
- Weil R, Israel A. Deciphering the pathway from the TCR to NF-kappaB. Cell Death Differ 2006. 13(5):826-833. [DOD] [CrossRef]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol 2002. 3(3):221-227. [DOD] [CrossRef]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005. 120(5):649-661. [DOD] [CrossRef]
- Bubici C, Papa S, Pham CG, Zazzeroni F, Franzoso G. The NF-kappaB-mediated control of ROS and JNK signaling. Histol Histopathol 2006. 21(1):69-80. [DOD]
- Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci U S A 2003. 100(25):15035-15040. [DOD] [CrossRef]
- Palmer E. Negative selection - clearing out the bad apples from the T-cell repertoire. Nat Rev Immunol 2003. 3:383-391. [DOD] [CrossRef]
- Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol 2003. 21:139-176. [DOD] [CrossRef]
- Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell 1987. 49:273-280. [DOD] [CrossRef]
- MacDonald HR, Schneider R, Lees RK, Howe RC, Acha-Orbea H, Festenstein H, Zinkernagel RM, Hengartner H. T cell receptor V beta use predicts reactivity and tolerance to Mlsa-encoded antigens. Nature 1988. 332:40-45. [DOD] [CrossRef]
- Kappler JW, Staerz UD, White J, Marrack P. Self tolerance eliminates T cells specific for Mls-modified products of the major histocompatibility complex. Nature 1988. 332:35-40. [DOD] [CrossRef]
- Kisielow P, Blüthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T cell receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature 1988. 333:742-746. [DOD] [CrossRef]
- Teh HS, Kisielow P, Scott B, Kishi H, Uematsu Y, Blüthmann H, von Boehmer H. Thymic major histocompatibility complex antigens and the alpha beta T cell receptor determine the CD4/CD8 phenotype of T cells. Nature 1988. 335:229-233. [DOD] [CrossRef]
- Smith KM, Olson DC, Hirose R, Hanahan D. Pancreatic gene expression in rare cells of thymic medulla: evidence for functional contribution to T cell tolerance. Int Immunol 1997. 9:1355-1365. [DOD] [CrossRef]
- Kyewski B, Derbinski J, Gotter J, Klein L. Promiscuous gene expression and central T-cell tolerance: more than meets the eye. Trends Immunol 2002. 23:364-371. [DOD] [CrossRef]
- The Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 1997. 17:399-403. [DOD] [CrossRef]
- Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N. Positional cloning of the APECED gene. Nat Genet 1997. 17:393-398. [DOD] [CrossRef]
- Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002. 298:1395-1401. [DOD] [CrossRef]
- Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kampe O, Eskelin P, Pelto-Huikko M, Peltonen L. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum Mol Genet 2002. 11:397-409. [DOD] [CrossRef]
- Webb S, Morris C, Sprent J. Extrathymic tolerance of mature T cells: Clonal elimination as a consequence of immunity. Cell 1990. 63:1249-1256. [DOD] [CrossRef]
- Rocha B, von Boehmer H. Peripheral selection of the T cell repertoire. Science 1991. 251:1225-1228. [DOD] [CrossRef]
- Kawabe Y, Ochi A. Programmed cell death and extrathymic reduction of Vbeta8+ CD4+ T cells in mice tolerant to Staphylococcus aureus enterotoxin B. Nature 1991. 349:245-247. [DOD] [CrossRef]
- Chen Y, Inobe J, Marks R, Gonnella P, Kuchroo VK, Weiner HL. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature 1995. 376:177-180. [DOD] [CrossRef]
- Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med 1996. 184:923-930. [DOD] [CrossRef]
- Liblau RS, Tisch R, Shokat K, Yang XD, Dumont N, Goodnow CC, McDevitt HO. Intravenous injection of soluble antigen induces thymic and peripheral T-cell apoptosis. Proc Natl Acad Sci USA 1996. 93:3031-3036. [DOD] [CrossRef]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992. 356:314-317. [DOD] [CrossRef]
- Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 1994. 76:969-976. [DOD] [CrossRef]
- Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 1995. 268:1347-1349. [DOD] [CrossRef]
- Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middelton LA, Lin AY, Strober W, Lenardo MJ, Puck JM. Dominant interfering Fas gene mutations. Cell 1995. 81:935-946. [DOD] [CrossRef]
- Lenardo MJ, Chan FK, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis - immune regulation in a dynamic and unpredictable environment. Annu Rev Immunol 1999. 17:221-253. [DOD] [CrossRef]
- Russell JH, Rush B, Weaver C, Wang R. Mature T cells of autoimmune lpr/lpr mice have a defect in antigen-stimulated suicide. Proc Natl Acad Sci USA 1993. 90:4409-4413. [DOD] [CrossRef]
- Singer GG, Abbas AK. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity 1994. 1:365-371. [DOD] [CrossRef]
- Zhou T, Bluethmann H, Zhang J, Edwards III CK, Mountz JD. Defective maintenance of T cell tolerance to a superantigen in MRL-lpr/lpr mice. J Exp Med 1992. 176:1063-1072. [DOD] [CrossRef]
- Sytwu HK, Liblau RL, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity 1996. 5:17-30. [DOD] [CrossRef]
- Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, Marrack PC. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity 1999. 10:735-744. [DOD] [CrossRef]
- Nguyen LT, McKall-Faienza K, Zakarian A, Speiser DE, Mak TW, Ohashi PS. TNF receptor 1 (TNFR1) and CD95 are not required for T cell deletion after virus infection but contribute to peptide-induced deletion under limited conditions. Eur J Immunol 2000. 30:683-688. [DOD] [CrossRef]
- Reich A, Korner H, Sedgwick JD, Pircher H. Immune down-regulation and peripheral deletion of CD8 T cells does not require TNF receptor-ligand interactions nor CD95. Eur J Immunol 2000. 30:678-682. [DOD] [CrossRef]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr, Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996. 85:549-561. [DOD] [CrossRef]
- Murga M, Fernandez-Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, Balomenos D, Vicario A, Carrera AC, Orkin SH, Greenberg ME, Zubiaga AM. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 2001. 15:959-970. [DOD] [CrossRef]
- Zhang Y, Schlossman SF, Edwards RA, Ou CN, Gu J, Wu MX. Impaired apoptosis, extended duration of immune responses, and a lupus-like autoimmune disease in IEX-1-transgenic mice. Proc Natl Acad Sci USA 2002. 99:878-883. [DOD]
- Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, Mathis D. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity 2005. 22:385-396. [DOD] [CrossRef]
- Decallonne B, van Etten E, Giulietti A, Casteels K, Overbergh L Bouillon, Mathieu C. Defect in activation-induced cell death in non-obese diabetic (NOD) T lymphocytes. J Autoimmun 2003. 20(3):219-226. [DOD] [CrossRef]
- Arreaza G, Salojin K, Yang W, Zhang J, Gill B, Mi QS, Gao JX, Meagher C, Cameron M, Delovitch TL. Deficient activation and resistance to activation-induced apoptosis of CD8+ T cells associated with defective peripheral tolerance in nonobese diabetic mice. J Autoimmun 2003. 107(2):103-115. [DOD]
- Nolsoe RL, Kristiansen OP, Larsen ZM, Johannesen J, Pociot F, Mandrup-Poulsen T. Complete mutation scan of the human Fas ligand gene: linkage studies in Type I diabetes mellitus families. Diabetologia 2003. 20(3):219-226. [DOD]
- Qin HY, Chaturvedi P, Singh B. In vivo apoptosis of diabetogenic T cells in NOD mice by IFN-gamma/TNF-alpha. Int Immunol 2004. 16(12):1723-1732. [DOD] [CrossRef]
- Papadopoulos K. Targeting the bcl-2 family in cancer therapy. Semin Oncol 2006. 33(4):449-456. [DOD] [CrossRef]
- Mathieu C, Waer M, Laureys J, Rutgeerts O, Bouillon R. Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia 1994. 37:552-558. [DOD]
This article has been cited by other articles:

|
Up-regulation of fas and fasL pro-apoptotic genes expression in type 1 diabetes patients after autologous haematopoietic stem cell transplantation
de Oliveira GL, Malmegrim KC, Ferreira AF, Tognon R, Kashima S, Couri CE, Covas DT, Voltarelli JC, de Castro FA
Clin Exp Immunol 2012. 168(3):291-302
|
|

|
T-cell death following immune activation is mediated by mitochondria-localized SARM
Panneerselvam P, Singh LP, Selvarajan V, Chng WJ, Ng SB, Tan NS, Ho B, Chen J, Ding JL
Cell Death Differ 2012. Epub
|
|

|
Immune profiling by multiple gene expression analysis in patients at-risk and with type 1 diabetes
Han D, Leyva CA, Matheson D, Mineo D, Messinger S, Blomberg BB, Hernandez A, Meneghini LF, Allende G, Skyler JS, Alejandro R, Pugliese A, Kenyon NS
Clin Immunol 2011. 139(3):290-301
|
|

|
Autoimmune thyroiditis, pernicious anaemia, vitiligo and scleroatrophic lichen in a boy with short-chain AcylCoA dehydrogenase deficiency
Stagi S, Gasperini S, Manoni C, Greco A, Funghini S, Donati A
Horm Res Paediatr 2010. 73(5):409-413
|
|

|
Gene therapy in diabetes
Wong MS, Hawthorne WJ, Manolios N
Self Nonself 2010. 1(3):165-175
|
|

|
Mitochondrial DNA variants in the pathogenesis of type 2 diabetes - relevance of asian population studies
Wang PW, Lin TK, Weng SW, Liou CW
Rev Diabet Stud 2009. 6(4):237-246
|
|

|
Mitochondrial nutrients improve immune dysfunction in the type 2 diabetic Goto-Kakizaki rats
Hao J, Shen W, Tian C, Liu Z, Ren J, Luo C, Long J, Sharman E, Liu J
J Cell Mol Med 2008. Epub ahead of print
|
|

|
BimEL-dependent apoptosis induced in peripheral blood lymphocytes with n-butyric acid is moderated by variation in expression of c-myc and p21(WAF1)
Kalousek I, Brodska B, Otevrelova P, Röselova P
Cell Biochem Funct 2008. 26(4):509-521
|
|

|
Molecular mechanisms of autoimmunity
Atassi MZ, Casali P
Autoimmunity 2008. 41(2):123-132
|
|

|
The Cross-Regulatory Relationship Between Human Dendritic and Regulatory T Cells and its Role in Type 1 Diabetes Mellitus
Chang HW, Chow YH, Chong P, Sia C
Rev Diabet Stud 2007. 4(2):68-76
|
|
|


)

