Diabetic Perspectives
| Rev Diabet Stud,
2006,
3(3):143-150 |
DOI 10.1900/RDS.2006.3.143 |
Advanced Diabetic Neuropathy: A Point of no Return?
Petr Boucek
Diabetes Centre, Institute for Clinical and Experimental Medicine, Videnska 1958/9, 14021 Prague 4, Czech Republic, e-mail: petr.boucek@ikem.cz.
Keywords: diabetic neuropathy, pathogenetic mechanisms and treatment, experimental and clinical trials
Abstract
Diabetic peripheral neuropathy is the most common complication of long-standing diabetes mellitus which frequently results in clinically significant morbidities e.g. pain, foot ulcers and amputations. During its natural course it progresses from initial functional changes to late, poorly reversible, structural changes. Various interconnected pathogenetic concepts of diabetic neuropathy have been proposed based on metabolic and vascular factors, mostly derived from long-term hyperglycemia. These pathogenetic mechanisms have been targeted in several experimental and clinical trials. This review summarizes available, mainly morphological data from interventions designed to halt the progression or achieve the reversal of established diabetic neuropathy, which include the recovery of normoglycemia by pancreas or islet transplantation, polyol pathway blockade by aldose reductase inhibitors, mitigation of oxidative stress by the use of antioxidants or correction of abnormalities in essential fatty acid metabolism. Unfortunately, to date, no treatment based on pathogenic considerations has shown clear positive effects and thus early institution of optimal glycemic control remains the only available measure with proven efficacy in preventing or halting progression of diabetic neuropathy. Further experimental and clinical research employing objective reproducible parameters is clearly needed. Novel non-invasive or minimally invasive methods e.g. corneal confocal microscopy or epidermal nerve fiber counts may represent potentially useful instruments for the objective assessment of nerve damage and monitoring of treatment effects.
Introduction
Diabetic peripheral neuropathy is the most common complication of long-standing diabetes mellitus (DM) and through its much-feared sequel, the diabetic foot, is responsible for most diabetes-related hospitalizations [1]. During its natural course it progresses from initial functional to late structural changes. The main morphological features of established neuropathy include a combination of demyelinization and axonal degeneration of myelinated fibers, degeneration with regeneration of unmyelinated fibers and endoneurial microangiopathy, with nerve fiber loss in its final stage [2]. Neuropathy frequently results in clinically significant morbidities, such as pain, loss of sensation, foot ulcers, gangrene and amputations.
Optimal metabolic control is the only available measure with proven efficacy in preventing or at least halting the progression of diabetic neuropathy [3]. However, to be effective it should be instituted at an early stage since, as is the case with other late complications of diabetes, the late phases of diabetic neuropathy are poorly reversible or even irreversible. Moreover, ample evidence of defective nerve regeneration in DM is available [4-6].
Apart from a brief review of pathogenetic and pathophysiological mechanisms involved in the progressive course of diabetic neuropathy (with a special emphasis on the problems of nerve regeneration) the main aims of the following text are: (i) to summarize available data on experimental and clinical interventions aiming to halt the progression or reverse the course of diabetic neuropathy and (ii) to highlight some novel objective instruments for the assessment of nerve damage and the monitoring of potential treatment effects.
This review will concentrate mainly on data derived from morphological examinations as a sensitive and reproducible method for assessing the severity of diabetic neuropathy [7, 8]. Conventional clinical scores of symptoms and signs, quantitative sensory testing and electrophysiological measurements are the methods which have been most frequently used to assess disease severity and efficacy of therapeutic interventions. However, in the advanced stages of neuropathy especially, when various types of responses to tests can frequently no longer be evoked, the utilization of objective quantitative morphometric parameters provides unique information. On the other hand, evidence of morphological improvement should be followed by proof of functional repair.
Current concepts of pathogenetic mechanisms of diabetic neuropathy
Chronic hyperglycemia represents the main causative factor involved in the pathogenesis of diabetic microvascular complications (retinopathy, nephropathy, and neuropathy). Nerve damage may be directly induced by the accumulation of intracellular glucose and its increased flux through the polyol pathway (with aldose reductase as the rate-limiting enzyme), the consequences of which include the generation of glycating sugars and advanced glycation end-products (AGE), enhanced oxidative damage and protein kinase C activation [9].
Apart from direct hyperglycemia-induced damage, ischemia caused indirectly from decrease in neurovascular flow almost certainly plays a role [10]. In this respect, the impairment of essential fatty acid synthesis and compromised vasoactive prostanoid production in diabetes may be a contributing factor [11]. Early changes in vascular function evolve into the progressive chronic endoneurial vascular damage seen in parallel to neurodegeneration in biopsy samples from diabetic patients [12]. Data from animal models of diabetes also indicate that deficiencies of neurotrophic factors and their receptors are involved in the development of diabetic neuropathy [1]. In this context, insulin and C-peptide deficiencies have been implicated as potential contributors to the generally more severe neuropathy of type 1 rather than that of type 2 diabetes [13]. There is also some evidence that autoimmune mechanisms may contribute to the development of symptomatic autonomic neuropathy in diabetic patients.
Problems with nerve regeneration in diabetes mellitus
Impairment of peripheral nerve regeneration in diabetes is suggested by numerous experimental studies. Regeneration following injury (crush, freeze or transection) was preferentially studied, even though this may not necessarily be representative of events subsequent to nerve damage caused by long-term diabetes. Regeneration was slowed after nerve injury in rat models of type 1 DM with alterations occurring in various components of the process. For example Wallerian degeneration, which is a precondition for normal regeneration following nerve injury, was delayed in the nerves of streptozotocin(STZ)-induced diabetic rats [14]. Both crush and transection of sciatic nerves in diabetic mice (STZ-induced) were followed by defective repopulation and maturation of axons [15], which was confirmed by various morphological and electrophysiological approaches.
Several putative mechanisms which were enumerated in recent reviews [4, 5] may be responsible for the decreased nerve regenerative capacity. They include microangiopathy with endoneurial hypoxia, excessive oxidative stress with overproduction of harmful reactive oxygen species, defects in macrophage function and inflammatory repair, reduced regenerative activity on the part of Schwann cells and impaired neuronal cell body response caused by deficient neurotrophic support [5].
Effects of restoration of normoglycemia and other therapeutic interventions
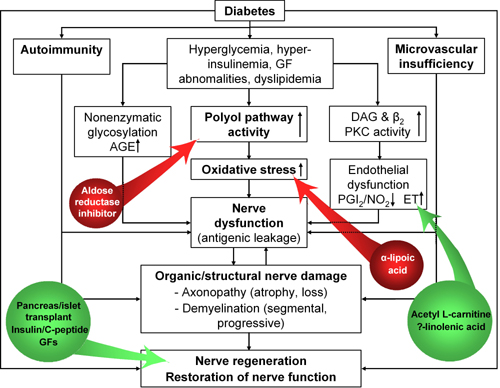
The interdependence of the hypothetic pathogenetic pathways involved provides the rationale for the use of various therapeutic strategies aimed at key initial abnormalities, i.e. chronic hyperglycemia and derived early defects [9]. Although reports on other treatment effects have been published, interventions for which morphological data are available include primarily restoration of normoglycemia by pancreas (or islet) transplantation and polyol pathway blockade by aldose reductase inhibition. Several other types of treatment based on putative pathogenetic mechanisms (e.g. with the antioxidant α-lipoic acid, γ-linolenic acid-containing oils to correct essential fatty acid metabolism, neurotrophic substances such insulin, C-peptide or nerve growth factor) have also been tried (Figure 1).
 |
 |
Figure 1. Therapeutic interventions based on hypothetic pathogenetic mechanisms of diabetic neuropathy. Interventions with inhibitory effects are marked in red, with mainly supportive effects in green. AGE: advanced glycation end products. GF: growth factor. DAG: diacylglycerol. PKC: protein kinase C. PG: prostaglandin. NO: nitric oxide. ET: endothelin (modified according to [16]). |
|
Experimental data
When considering the preventive and restorative effects of individual treatments, it is important to bear in mind that despite the presence of significant electrophysiological abnormalities some common diabetic rat models (e.g. STZ-induced diabetic rats) may not exhibit the advanced structural changes and fiber loss seen in human neuropathy [11]. Moreover, despite similar hyperglycemic levels, differences exist between models of type 1 and type 2 DM in their expression of morphologic changes [17].
Most recently, Kennedy and Zochodne described physiological and structural features of diabetic neuropathy which closely model human disease in a long-term murine model of STZ-induced DM. A subset of these diabetic mice which spontaneously regained islet function and attained near-normoglycemia demonstrated significant electrophysiological improvement, less myelin thinning and clear epidermal and sweat gland reinnervation. However, no recovery of sensory neuron loss in dorsal root ganglia was observed, which underscores the importance of early preventive measures to protect sensory neuron populations [18].
The effect of pancreatic islet cell allotransplantation with restoration of normoglycemia on the development of diabetic neuropathy was examined morphometrically by Sima et al. with light and electron microscopy in STZ-induced diabetic rats. Successful allotransplantation with return to euglycemia completely prevented axonal atrophy and the characteristic nodal and paranodal structural abnormalities in diabetic nerves, as well as the typical slowing of nerve conduction velocity [19]. Orloff et al. reported that pancreatic islet transplantation prevented diabetic neuropathy (5 nerve lesions confirmed by electron microscopy) in syngeneic alloxan-diabetic rats for the first 6 months. When hyperglycemia recurred, nerve lesions similar in severity to those in untreated diabetic rats developed [20].
On the other hand, as reported by the same group, whole-pancreas transplants produced long-term metabolic control of DM, prevented the development and progression of diabetic nerve lesions throughout a 2-year study period and reversed all the lesions in both sciatic and testicular nerves, even when carried out late in the course of DM. There was complete reversal of the nerve lesions when pancreas transplantation (PT) was performed within 15 months of the onset of DM [21]. In a study in alloxan-diabetic Lewis rats by Scott et al. [22], PT had a beneficial effect on the axonal cross-sectional area of myelinated nerve fibers and the relative percentages of the various sizes of nerve fibers, especially when performed in early diabetes. The effects in late diabetes were less spectacular and PT did not prove beneficial to unmyelinated fiber densities. The findings suggest that pancreas (or islet) transplantation with establishment of euglycemia may represent an effective therapeutic approach to the prevention and even reversal of early experimental diabetic neuropathy.
The effect of treatment with aldose reductase inhibitors (ARIs) has been extensively studied in various rodent models of diabetic neuropathy. As reported by Sima et al., ponalrestat treatment of the spontaneously diabetic BB-rat (type 1 DM model) for 6 months achieved a partial but significant prevention of nerve conduction slowing, axoglial dysjunction, and axonal degenerative changes. A more than threefold increase in regenerating nerve fibers was also observed. In the opinion of the authors additional pathogenetic mechanisms besides polyol-pathway activation or, alternatively, insufficient ARI dosage could be responsible for the incomplete prevention of neuropathy [23]. In a study by Yahigashi et al., structural analysis of the sural nerve of STZ-induced diabetic rats indicated that ponalrestat treatment for 28 weeks had significant preventive effects on the reduction in myelinated nerve fiber size and fiber occupancy. Axon-fiber size ratio was also preserved in the ponalrestat-treated group [24].
The effect of tolrestat treatment on nerve regeneration after sciatic nerve crush injury was studied by Terada et al. [25] in STZ-induced diabetic rats. The significant decrease in the density of myelinated nerve fibers and their number per fascicle seen in untreated diabetic rats were prevented by tolrestat. Following axotomy, impaired nerve fiber regeneration in diabetic BB/W rats was observed in a study by Kamijo et al. [26]. It was associated with a marked delay in preceding Wallerian degeneration and decreased phagocytic activity by macrophages, changes not demonstrated in ARI-treated diabetic rats. Thus, the blockade of aldose reductase activity by ARIs resulted not only in attenuation of several metabolic, vascular or physiologic defects, but in some cases also produced morphological improvements.
Treatments with various antioxidants (e.g. α-lipoic acid, acetylcysteine and α-tocopherol) [27-29], γ-linolenic acid-containing oils [30] or acetyl-L-carnitine [31] have also been shown to produce some benefits on nerve function and even morphology in experimental diabetic models. However, a more detailed description of the diverse range of interventions would far exceed the scope of this text.
The fact that the aforementioned difference in the expression of neuropathy results in more severe neuropathic deficits in type 1 than in type 2 diabetic models in spite of similar levels of hyperglycemia seems to suggest a direct pathogenetic role of insulin (and/or C-peptide) deficiency [32]. In support of this concept, eight months of C-peptide replacement in a study by Sima et al. partially prevented chronic nerve conduction defects and totally prevented axoglial dysjunction and paranodal demyelination in diabetic BB/Wor rats. C-peptide treatment from 5 to 8 months after DM onset showed an improvement in nerve conduction velocity (NCV) and axoglial dysjunction, normalization of paranodal demyelination, repair of axonal degeneration and a fourfold increase in nerve fiber regeneration [33].
Intrathecal delivery of low doses of insulin (or insulin-like growth factor-1), insufficient to reduce glycemia, reversed atrophy in myelinated sensory axons in the sural nerve and improved motor and sensory nerve conduction velocity in STZ-induced diabetic rats [34]. Thus, defects in direct insulin signaling on peripheral neurons may play a role in the development of diabetic neuropathy.
Clinical data
Pancreas transplantation currently represents the only clinical method, which reliably re-establishes long-term normoglycemia in diabetic subjects (despite the major breakthrough accomplished by the introduction of the Edmonton protocol, islet transplantation still lags behind in this respect). PT is most frequently performed in type 1 diabetic patients in advanced stages of diabetic nephropathy and with other microvascular complications. Diabetic neuropathy, involving motor, sensory and autonomic nerves to a similar extent, is very common and severe in these patients [35, 36].
The long-term effects of re-established normoglycemia have been studied both in non-uremic patients receiving a pancreas transplant alone (PTA) and uremic patients given simultaneous pancreas and kidney transplant (SPK). Allen et al. followed 44 patients after a successful SPK for up to 8 years. Two distinct patterns of neurological recovery were observed. Rapid, initial recovery in NCV was followed by stabilization, whereas action potential amplitudes recovered in a slow, monophasic pattern, which persisted for the duration of the study [37].
The largest and longest experience was reported from the transplant centre of the University of Minnesota, where Navarro et al. followed patients with a functioning pancreas graft for up to 10 years. NCV of large motor and sensory nerve fibers increased significantly, whereas the amplitude of muscle action potential (MAP) did so only in nerves mildly involved, such as those of the upper extremity. The amplitude of nerve action potential (NAP) increased in the median and sural nerves. As interpreted by the authors, improvement of NCV after short-term treatment may represent functional rather than structural changes in peripheral nerves; later improvement may be due to remyelination and enlargement of atrophied axons. The increase in amplitude of MAP and NAP after PT was an indirect indication of the presence of new regenerating fibers [38]. On the other hand, most studies have failed to detect any beneficial effect of PT on cardiorespiratory reflex testing, i.e. on functions mediated by small nerve fibers [39, 40].
To date, no systematic study has documented nerve regeneration directly through morphological examination of peripheral nerves following a successful PT. Nonetheless, in a single case report Beggs et al. [41] identified signs suggestive of tissue repair and regeneration of some nerve components in sequential sural nerve biopsies following PTA in a type 1 diabetic patient. These included a reduction in the frequency of abnormal endoneurial capillary pericytes and in myelinated nerve fiber abnormalities characteristic of degeneration as well as an increase in those associated with fiber regeneration, doubling of myelinated nerve fiber sprouts and a two-fold increase in unmyelinated axons.
Since the invasive nature of sural nerve biopsy precludes its wider and more frequent application, Kennedy et al. have suggested that quantification of epidermal nerve fibers stained by the sensitive panaxonal marker, protein gene product (PGP) 9.5, in lower limb skin biopsy samples could be used as a follow-up measure of neuropathy after PT [42]. While extensive depletion of epidermal nerve fibers was present in baseline biopsies, their increased numbers could be demonstrated in rebiopsies of 4 out of 10 patients following PT [43]. A systematic follow-up study of similar design has been started at our centre in collaboration with the Department of Neurology of the University of Würzburg, Germany (Prof. C. Sommer). However, preliminary results in 16 patients (1 PTA, 15 SPK) after a mean of 18 months of post-transplant normoglycemia indicate that such a period is insufficient to document a significant increase in the mean number of epidermal nerve fibers in thigh and calf biopsy samples [44].
In summary, data from patients undergoing PT seem to suggest some nerve regenerative tendency mainly in less severe cases. However, no marked effect on neuropathic deficits has been demonstrated and the risk of neuropathic diabetic foot complications persists in the post-transplant period. Thus, additional types of interventions apart from the abolishment of the diabetic metabolic milieu by successful pancreatic transplantion would certainly be of interest.
Various types of ARIs have been used in clinical trials of diabetic neuropathy including trials where structural changes have been assessed. In a 1-year double-blind, placebo-controlled trial by Sima et al. 10 patients with neuropathy were treated with sorbinil (250mg/day).The sorbinil-treated patients demonstrated a decrease in nerve sorbitol content and an increase in the number of myelinated fibers per unit of cross-sectional area of nerve in follow-up sural nerve biopsies. They also showed a quantitative improvement in terms of the degree of paranodal demyelination, segmental demyelination, and myelin wrinkling. The increase in the number of fibers was accompanied by electrophysiological and clinical evidence of improved nerve function [45].
Changes in nerve morphology were also assessed in patients with mild to moderate peripheral polyneuropathy in a 1-year, randomized, placebo-controlled, double-blind, multiple-dose trial of zenarestat by Greene et al. Contralateral sural nerve biopsies were obtained at 6 weeks and at the study's completion for nerve sorbitol measurement and computer-assisted light morphometry to determine myelinated nerve fiber density in serial bilateral sural nerve biopsies. Doses of zenarestat producing >80% sorbitol suppression were associated with a significant increase in the density of sural nerve myelinated fibers especially those of small-diameter (<5 micron) [46]. Thus, morphologic evaluation of sural nerve biopsies obtained after ARI treatment suggests that these biochemically effective compounds ameliorate clinically relevant structural lesions in patients with diabetic neuropathy [47]. However, in spite of the fact that some ARI compounds have yielded positive results in experimental and some clinical trials, several factors, mainly poor tolerance and toxicity, have so far precluded their introduction into clinical practice.
Though both improvement in neuropathic symptoms and deficits have been demonstrated with short-term treatment using α-lipoic [48] or γ-linolenic acid [49], changes in nerve morphology were not assessed in any of these trials. Moreover, it has been disclosed recently that a pivotal multi-center trial of oral treatment of diabetic polyneuropathy with α-lipoic acid (NATHAN I) combining clinical and neurophysiological assessments failed to confirm its long-term efficacy [50]. On the other hand, two studies demonstrated improvements in symptoms, vibration perception as well as sural nerve morphometry in patients with chronic diabetic neuropathy following 1-year treatment with two doses of acetyl-L-carnitine [51].
Several lines of evidence suggest that deficiency of neurotrophic factors may play a role in the pathogenesis of diabetic peripheral neuropathy. However, high hopes for a role for recombinant human nerve growth factor (rhNGF) in the treatment of diabetic neuropathy were dashed by its well-known failure in a phase III clinical trial conducted in the USA [52]. Another clinical study of diabetic polyneuropathy using the brain-derived neurotrophic factor (rhBDNF) was also unsuccessful [53]. Hypothetically, the failure of the growth factor studies may in part be due to the use of inappropriate measures which could have failed to register repair of a specific nerve fiber population such as fibers of small caliber. There have been no reports concerning the effect of rhNGF or rhBDNF on human peripheral nerve morphology.
Potential new avenues and future prospects
Since definitive reversal of advanced structural neuropathy has remained an elusive goal in past clinical research, a consensus has emerged concerning the optimal timing of interventions aimed at improving or at least halting the progression of neuropathy. Interventions must be initiated at much earlier stages when the threshold of irreversibility has not yet been reached [54]. To identify this time point correctly new appropriate and widely utilizable markers should be introduced similar to microalbuminuria in the case of nephropathy, which has proved to be a valuable means of detecting the stage when it is still potentially reversible. Such simple and easily available markers of the phase when diabetic neuropathy can still be reversed are currently lacking. Although several parameters of quantitative sensory and electrophysiological testing (e.g. vibration perception thresholds or nerve potential amplitudes) correlate closely with morphological findings such as nerve fiber density and may identify patients amenable to therapy [55], their widespread use for the purpose of establishing reversibility thresholds remains questionable.
An interesting new approach to the accurate definition of nerve fiber damage and repair has been recently proposed by Malik et al. from Manchester [56]. The authors have used confocal microscopy, a rapid, non-invasive in vivo clinical examination technique, to assess nerve fiber density, length and branch density in Bowman’s layer of the cornea. These measurements were reduced in diabetic patients in comparison with control subjects, with a tendency towards a greater reduction as the severity of neuropathy increased. Thus, if confirmed by further investigations, corneal confocal microscopy may possibly act as a surrogate measure of somatic neuropathy in diabetic patients. It could be used to define the severity of neuropathy and record eventual nerve regeneration in clinical trials of human diabetic neuropathy. In a recent pilot study, improvement in nerve fiber density as a sign of early neural regeneration was detected by this method within 6-12 months of pancreas transplantation with sustained euglycemia [57].
The minimally invasive method mentioned above for the quantitation of epidermal nerve fibers in skin biopsies [58, 59] and blisters [60] is being adopted by several groups as a useful instrument for the morphological assessment of diabetic neuropathy. Recently, a new cutaneous nerve regeneration model using a capsaicin dressing was developed and validated in healthy controls and in diabetic subjects with and without neuropathy [61]. The authors showed that topical capsaicin application produced a uniform epidermal nerve fiber injury and that rates of regeneration of epidermal nerve fibers in subsequent cutaneous biopsies were reduced even in diabetic patients without evidence of neuropathy. They concluded that this procedure offered an efficient strategy to study nerve regeneration in diabetic subjects and that the duration of such trials could probably be shortened significantly.
Conclusions
In spite of the plethora of pathogenetic concepts based on extensive experimental research there is still a lack of effective measures derived from successful clinical trials to reverse the course of established diabetic neuropathy. Consequently, prevention of progression through close long-term glycemic control remains the only available effective clinical tool. Future efforts should concentrate on further defining and refining strategies for the conduct of clinical trials and evaluation of treatment effects [62] in order to translate what are at present little more than mere pathogenetic hypotheses into solid clinical knowledge.
Acknowledgments:
This work was supported by grant NR7929-4, Internal Grant Agency, Czech Ministry of Health.
References
- Vinik AI, Park TS, Stansberry KB, Pittenger GL. Diabetic neuropathies. Diabetologia 2000. 43:957-973. [DOD] [CrossRef]
- Malik RA, Tesfaye S, Newrick PG, Walker D, Rajbhandari SM, Siddique I, Sharma AK, Boulton AJ, King RH, Thomas PK, Ward JD. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia 2005. 48:578-585. [DOD] [CrossRef]
- DCCT Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Int Med 1995. 122(8):561-568. [DOD]
- Yasuda H, Terada M, Maeda K, Kogawa S, Sanada M, Haneda M, Kashiwagi A, Kikkawa R. Diabetic neuropathy and nerve regeneration. Prog Neurobiol 2003. 69(4):229-285. [DOD] [CrossRef]
- Kennedy JM, Zochodne DW. Impaired peripheral nerve regeneration in diabetes mellitus. J Peripher Nerv Syst 2005. 10:144-157. [DOD] [CrossRef]
- Apfel SC. Nerve regeneration in diabetic neuropathy. Diabet Obes Metab 1999. 1:3-11. [DOD] [CrossRef]
- Sima AA. Structure-function interactions in the therapeutic response of diabetic neuropathy. J Diabetes Complications 1992. 6:64-68. [DOD] [CrossRef]
- Sima AA. The reproducibility and sensitivity of sural nerve morphometry in the assessment of diabetic peripheral polyneuropathy. Diabetologia 1992. 35:560-569. [DOD] [CrossRef]
- Stevens MJ, Feldman EL, Thomas T, Greene DA. Pathogenesis of diabetic neuropathy. In: Veves A (ed). Clinical management of diabetic neuropathy. Humana Press, Totowa, NJ, 1998. Chapter 2, pp. 13-48. [DOD]
- Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA 2002. 288:2579-2588. [DOD] [CrossRef]
- Sima AA, Sugimoto K. Experimental diabetic neuropathy: an update. Diabetologia 1999. 42:773-788. [DOD] [CrossRef]
- Cameron NE, Eaton SEM, Cotter MA, Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 2001. 44:1973-1988. [DOD] [CrossRef]
- Sima AA, Pierson CR. Metabolic alterations in experimental models. In: Gries FA, Cameron NE, Low PA, Ziegler D (eds). Textbook of diabetic neuropathy. Thieme, Stuttgart, 2003. pp. 96-105. [DOD]
- Terada M, Yasuda H, Kikkawa R. Delayed Wallerian degeneration and increased neurofilament phosphorylation in sciatic nerves of rats with streptozotocin-induced diabetes. J Neurol Sci 1998. 155:23-30. [DOD] [CrossRef]
- Kennedy JM, Zochodne DW. The regenerative deficit of peripheral nerves in experimental diabetes: its extent, timing and possible mechanisms. Brain 2000. 123:2118-2129. [DOD] [CrossRef]
- Vinik AI, Pittenger GL, Barlow P, Mehrabyan A. Diabetic neuropathies: an overview of clinical aspects, pathogenesis and treatment. In: LeRoith D, Taylor SI, Olefsky JM (eds). Diabetes mellitus: a fundamental and clinical text. 3rd Edition, Lippincott, Philadelphia 2004. [DOD]
- Sima AA, Zhang W, Xu G, Sugimoto K, Guberski D, Yorek MA. A comparison of diabetic polyneuropathy in type II diabetic BBZDR/Wor rats and in type I diabetic BB/wor rats. Diabetologia 2000. 43:786-793. [DOD] [CrossRef]
- Kennedy JM, Zochodne DW. Experimental diabetic neuropathy with spontaneous recovery. Is there irreparable damage? Diabetes 2005. 54:830-837. [DOD]
- Sima AA, Zhang WX, Tai J, Tze WJ, Nathaniel V. Diabetic neuropathy in STZ-induced diabetic rat and effect of allogenic islet cell transplantation. Morphometric analysis. Diabetes 1988. 37:1129-1136. [DOD]
- Orloff MJ, Macedo A, Greenleaf GE. Effect of pancreas transplantation on diabetic somatic neuropathy. Surgery 1988. 104:437-444. [DOD]
- Orloff MJ, Greenleaf GE, Girard B. Reversal of diabetic somatic neuropathy by whole-pancreas transplantation. Surgery 1990. 108:179-189. [DOD]
- Scott MH, Lee S, D'Silva M, Chang CM, Allen J, Yancey D, Moossa AR. Effect of pancreas allografts on the ultrastructure of sciatic nerves in diabetic rats. Microsurgery 1990. 11:152-161. [DOD]
- Sima AA, Prashar A, Zhang WX, Chakrabarti S, Greene DA. Preventive effect of long-term aldose reductase inhibition (ponalrestat) on nerve conduction and sural nerve structure in the spontaneously diabetic Bio-Breeding rat. J Clin Invest 1990. 85:1410-1420. [DOD]
- Yagihashi S, Kamijo M, Ido Y, Mirrlees D. Effects of long-term aldose reductase inhibition on development of experimental diabetic neuropathy: ultrastructural and morphometric studies of sural nerve in streptozotocin-induced diabetic rats. Diabetes 1990. 39:690-696. [DOD]
- Terada M, Yasuda H, Kikkawa R, Shigeta Y. Tolrestat improves nerve regeneration after crush injury in streptozotocin-induced diabetic rats. Metabolism 1996. 45:1189-1195. [DOD] [CrossRef]
- Kamijo M, Merry AC, Cherian PV, Zkras G, Sima AA. Nerve fibre regeneration following axotomy in the diabetic BB/W-rat. The effect of ARI treatment. J Diabetes Complications 1996. 10:183-191. [DOD] [CrossRef]
- Nagamatsu M, Nickander KK, Schmelzer JD. Lipoic acid improves nerve blood flow, reduces oxidative stress and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care 1995. 18:1160-1167. [DOD]
- Love A, Cotter MA, Cameron NE. Effects of the sulphydryl donor N-acetyl-L-cysteine on nerve conduction, perfusion, maturation and regeneration following freeze damage in diabetic rats. Eur J Clin Invest 1996. 26:698-706. [DOD]
- Love A, Cotter MA, Cameron NE. Effects of alpha tocopherol on nerve conduction velocity and regeneration following freeze lesion in immature diabetic rats. Naunyn Schmiedebergs Arch Pharmacol 1997. 355:126-130. [DOD] [CrossRef]
- Cameron NE, Cotter MA. Comparison of the effects of ascorbyl gamma-linolenic acid and gamma-linolenic acid in the correction of neurovascular deficits in diabetic rats. Diabetologia 1996. 39:1047-1054. [DOD]
- Sima AA, Ristic H, Merry A, Kamijo M, Lattimer SA, Stevens MJ, Greene DA. Primary preventive and secondary interventionary effects of acetyl-L-carnitine on diabetic neuropathy in the bio-breeding Worcester rat. J Clin Invest 1996. 97:1900-1907. [DOD]
- Sima AA. Pathological mechanisms involved in diabetic neuropathy: can we slow the process? Curr Opin Investig Drugs 2006. 7:324-327. [DOD]
- Sima AA, Zhang W, Sugimoto K, Henry D, Li Z, Wahren J, Grunberger G. C-peptide prevents and improves chronic type 1 diabetic polyneuropathy in the BB/Wor rat. Diabetologia 2001. 44:889-897. [DOD] [CrossRef]
- Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes 2004. 53:1824-1830. [DOD]
- Kennedy WR, Navarro X, Sutherland DE. Neuropathy profile of diabetic patients in a pancreas transplantation program. Neurology 1995. 45:773-780. [DOD]
- Hathaway DK, Cashion AK, Milstead EJ, Winsett RP, Cowan PA, Wicks MN, Gaber AO. Autonomic dysregulation in patients awaiting kidney transplantation. Am J Kidney Dis 1998. 32:221-229. [DOD]
- Allen RD, Al-Harbi IS, Morris JG, Clouston PD, O'Connell PJ, Chapman JR, Nankivell BJ. Diabetic neuropathy after pancreas transplantation: determinants of recovery. Transplantation 1997. 63:830-838. [DOD] [CrossRef]
- Navarro X, Sutherland DE, Kennedy WR. Long-term effects of pancreatic transplantation on diabetic neuropathy. Ann Neurol 1997. 42:727-736. [DOD] [CrossRef]
- Ziegler D. Glycemic control. In: Gries FA, Cameron NE, Low PA, Ziegler D (eds). Textbook of diabetic neuropathy. Thieme, Stuttgart, 2003. pp. 91-96. [DOD]
- Boucek P, Saudek F, Adamec M, Janousek L, Koznarova R, Havrdova T, Skibova J. Spectral analysis of heart variation following simultaneous pancreas and kidney transplantation. Transplant Proc 2003. 35:1494-1498. [DOD] [CrossRef]
- Beggs JL, Johnson PC, Olafsen AG, Cleary CP, Watkins CJ, Targovnik JH. Signs of nerve regeneration and repair following pancreas transplantation in an insulin-dependent diabetic with neuropathy. Clin Transplant 1990. 4:133-141. [DOD]
- Kennedy WR, Wendelschafer-Crabb G, Johnson T. Quantitation of epidermal nerves in diabetic neuropathy. Neurology 1996. 47:1042-1048. [DOD]
- Navarro X, Kennedy WR. Effects of pancreas transplantation on secondary complications of diabetes neuropathy. In: Gruessner RW, Sutherland DE (eds). Transplantation of the pancreas. Springer, New York, 2004. pp. 483-496. [DOD]
- Boucek P, Havrdova T, Voska L, Lodererova A, Hejna A, Saudek F, Lipar K, Janousek L, Adamec M, Sommer C. Epidermal nerve fibre depletion in simultaneous pancreas and kidney transplant patients: no change at follow-up after 18 months of normoglycemia. In: 16th Annual Neurodiab Meeting. Ystad, Sweden, 2006. [DOD]
- Sima AA, Bril V, Nathaniel V, McEwen TA, Brown MB, Lattimer SA, Greene DA. Regeneration and repair of myelinated fibers in sural-nerve biopsy specimens from patients with diabetic neuropathy treated with sorbinil. N Engl J Med 1988. 319:548-555. [DOD]
- Greene DA, Arezzo JC, Brown MB. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology 1999. 53:580-591. [DOD]
- Greene D. Effects of aldose reductase inhibitors on the progression of nerve fiber damage in diabetic neuropathy. J Diabetes Complications 1992. 6:35-38. [DOD] [CrossRef]
- Ziegler D, Reljanovic N, Mehnert H, Gries FA. alpha-lipoic acid in the treatment of diabetic polyneuropathy in Germany: current evidence from clinical trials. Exp Clin Endocrinol Diabetes 1999. 107:421-430. [DOD]
- Gamma-Linolenic Acid Multicenter Trial Group. Treatment of diabetic neuropathy with gamma-linolenic acid. Diabetes Care 1993. 16:8-15. [DOD]
- Ziegler D. Antioxidant treatment in diabetic polyneuropathy - update 2006. In: 16th Annual Neurodiab Meeting. Ystad, Sweden, 2006. [DOD]
- Sima AA, Calvani M, Mehra M, Amato A. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Acetyl-L-Carnitine Study Group. Diabetes Care 2005. 28(1):89-94. [DOD]
- Apfel SC, Schwartz S, Adornato BT, Freeman R, Biton V, Rendell M, Vinik A, Giuliani M, Stevens JC, Barbano R, Dyck PJ. Efficacy and safety of recombinant human nerve growth factor in patients with diabetic polyneuropathy. JAMA 2000. 284:2215-2221. [DOD] [CrossRef]
- Wellmer A, Misra VP, Sharief MK, Kopelman PG, Anand P. A double-blind placebo-controlled clinical trial of recombinant human brain-derived neurotrophic factor (rhBDNF) in diabetic polyneuropathy. J Peripher Nerv Syst 2001. 6:204-210. [DOD] [CrossRef]
- Pfeifer MA, Schumer MP. Clinical trials of diabetic neuropathy: past, present, and future. Diabetes 1995. 44:1355-1361. [DOD]
- Vinik AI, Bril V, Litchy WJ, Price KL, Bastyr EJ. Sural sensory action potential identifies diabetic peripheral neuropathy responders to therapy. MBBQ Study Group. Muscle Nerve 2005. 32:619-625. [DOD] [CrossRef]
- Malik RA, Kallinikos P, Abbott CA, van Schie CH, Morgan P, Efron N, Boulton AJ. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia 2003. 46:683-688. [DOD]
- Mehra S, Tavakoli M, Palmer A, Tavakoli A, Pararajasingam R, Parrott NR, Malik R, Augustine T. Early neural regeneration after pancreas transplantation detected by corneal confocal microscopy: a pilot study. Am J Transplant 2006. 82(Suppl 2):A118. [DOD]
- Levy DM, Terenghi G, Gu XH, Abraham RR, Springall DR, Polak JM. Immunohistochemical measurements of nerves and neuropeptides in diabetic skin: relationship to tests of neurological function. Diabetologia 1992. 35:889-897. [DOD] [CrossRef]
- McCarthy BG, Hsieh ST, Stocks A, Hauer P, Macko C, Cornblath DR, Griffin JW, McArthur JC. Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology 1995. 45:1848-1855. [DOD]
- Kennedy WR, Nolano M, Wendelschafer-Crabb G, Johnson T, Tamura E. A skin blister method to study epidermal nerves in peripheral nerve disease. Muscle Nerve 1999. 22:360-371. [DOD] [CrossRef]
- Polydefkis M, Hauer P, Sheth S, Sirdofsky M, Griffin JW, McArthur JC. The time course of epidermal nerve fibre regeneration: studies in normal controls and in people with diabetes, with and without neuropathy. Brain 2004. 127:1606-1615. [DOD] [CrossRef]
- Luft D, Ziegler D. Evaluation of drug effects. In: Gries FA, Cameron NE, Low PA, Ziegler D (eds). Textbook of diabetic neuropathy. Thieme, Stuttgart, 2003. pp. 313-327. [DOD]
This article has been cited by other articles:

|
Neuroprotective action of Ginkgo biloba on the enteric nervous system of diabetic rats
da Silva GG, Zanoni JN, Buttow NC
World J Gastroenterol 2011. 17(7):898-905
|
|
|
Relationship between neural alteration and perineural invasion in pancreatic cancer patients with hyperglycemia
Li J, Ma Q, Liu H, Guo K, Li F, Li W, Han L, Wang F, Wu E
PLoS One 2011. 6(2):e17385
|
|

|
Ranirestat: A Selective Aldose Reductase Inhibitor for Diabetic Sensorimotor Polyneuropathy
Vo T, Marcus KB
J Pharm Technol 2008. 24(6):340-344
|
|

|
Hyperglycemia promotes the perineural invasion in pancreatic cancer
Li J, Ma Q
Med Hypotheses 2008. 71(3):386-389
|
|

|
Antioxidant status in type 2 diabetic neuropathy
Mahmood IH, Al Neaimy KS
Bahrain Med Bull 2008. 30(1):12-15
|
|

|
The diabetic foot
Andersen CA, Roukis TS
Surg Clin North Am 2007. 87(5):1149-77
|
|
|


)

