Review
| Rev Diabet Stud,
2008,
5(1):13-24 |
DOI 10.1900/RDS.2008.5.13 |
The Role of Skeletal Muscle Sphingolipids in the Development of Insulin Resistance
Marek Straczkowski, Irina Kowalska
Department of Endocrinology, Diabetology and Internal Medicine, Medical University of Bialystok, Poland
Address correspondence to: Marek Straczkowski, e-mail: mstraczkowski@poczta.onet.pl
Manuscript submitted May 22, 2008; accepted May 29, 2008.
Keywords: type 2 diabetes, insulin resistance, skeletal muscle, lipids, ceramide, obesity, free fatty acids
Abstract
Insulin resistance is an important risk factor for type 2 diabetes, obesity, cardiovascular disease, polycystic ovary syndrome and other diseases. The most important stage in the development of insulin resistance is impairment of insulin-stimulated skeletal muscle glucose uptake. There is evidence that intramyocellular lipids might be responsible for this process through inhibition of insulin signaling. One of the important intracellular lipid pools is associated with the sphingomyelin signaling pathway. The second messenger in this pathway is ceramide. In vitro data indicate that ceramide inhibits insulin signaling, mainly through inactivation of protein kinase B. In vivo data suggest that ceramide accumulation within muscle cells might be associated with the development of insulin resistance. In this review, we discuss both in vitro and in vivo evidence for the role of muscle ceramide in the impairment of insulin action with particular focus on the question whether findings from animal studies are applicable to humans. We describe problems that are unresolved so far and topics of potential interest for future research.
Introduction
Insulin resistance, defined as an impaired metabolic response to insulin, is an important risk factor for type 2 diabetes, obesity, cardiovascular disease, polycystic ovary syndrome and other diseases [1, 2]. Skeletal muscle is mainly responsible for insulin-stimulated glucose uptake. Decreased skeletal muscle glucose uptake in response to insulin is a crucial step in the development of insulin resistance [3]. There are data that appear to indicate that impairment of insulin action might be related to intramyocellular lipid accumulation [4-8]. One of the important intracellular lipid pools is associated with the sphingomyelin signaling pathway. The second messenger in this pathway is ceramide [9]. The therapeutic potential of modulating the sphingomyelin signaling pathway has attracted increased interest in recent years [9]. Some review articles provide a good introduction to the role of the sphingomyelin signaling pathway and ceramide in insulin resistance [10-12]. In the present review, after describing the mechanisms of free fatty acid-induced insulin resistance in general terms, we discuss both in vitro and in vivo evidence of the role played by muscle ceramide in impairment of insulin action. In this context, we particularly focus on the question whether findings from animal studies are also applicable to humans.
Mechanisms of free fatty acid-induced insulin resistance
Increased delivery of free fatty acids (FFA) to peripheral tissues is one of the factors linking obesity with insulin resistance [13]. Evidence about the crucial role of FFA availability for impairment of insulin-stimulated muscle glucose uptake is available from both experimental and clinical studies.
Muscle-specific insulin resistance was detected in mice with muscle-specific overexpression of lipoprotein lipase [14]. Similarly, muscle-specific overexpression of FAT/CD36 in mice led to the development of insulin resistance, despite reduced circulating FFA and triacylglycerol (TAG) levels [15]. In contrast, mice with a null mutation of the FAT/CD36 gene had low serum glucose together with high FFA and TAG [16]. Inactivation of fatty acid transport protein 1 (FATP1) also protected mice from fat-induced insulin resistance, without changing body weight [17].
In humans, acute elevation of plasma FFA concentrations might be achieved through Intralipid/heparin infusion. Intralipid is a well-known lipid emulsion used for supplemental nutrition and diagnostic techniques. Such infusion results in a decrease in whole-body insulin-stimulated glucose uptake by 25-50% and this effect appears 3-4 hours after the beginning of the procedure [8, 18-21]. Initially, it was hypothesized that an increase in FFA would lead to an increase in their oxidation, thus reducing oxidation of glucose through the so-called "Randle cycle" [22]. However, it was demonstrated that fat oxidation was decreased in insulin-resistant individuals [23, 24]. AMP-activated protein kinase, an enzyme acting as 'energy sensor' within the cells, acts through inhibition of malonyl-CoA, thus enhancing muscle mitochondrial fatty acid β-oxidation and increasing insulin sensitivity [25]. Studies combining clamp techniques with indirect calorimetry, lipid infusion and nuclear magnetic resonance spectroscopy revealed that the initial step in the development of FFA-induced insulin resistance was impairment of glucose transport and phosphorylation in muscle cells. This was indicated by a decreased muscle glucose-6-phosphate level, with a subsequent decrease in both glucose oxidation and muscle glycogen synthesis [18]. These effects of FFA might be observed even at their physiological concentrations [26]. The decrease in glucose transport and phosphorylation is present in subjects with obesity [27] and a positive family history of type 2 diabetes [28]. Measurement of intracellular glucose in muscle revealed that impaired glucose transport, and not phosphorylation, is the primary abnormality in the development of lipid-induced insulin resistance in muscles [29].
The mechanism, by which FFA decrease insulin-stimulated glucose uptake is directly associated with an inhibition of insulin signaling within muscle cells (Figure 1). Lipid infusion abolished an increase in insulin receptor substrate 1 (IRS-1)-associated phosphatydiloinositol 3-kinase (PI3K) activity in response to insulin [29]. This effect, together with impaired Akt serine phosphorylation, is observed even within the physiological range of plasma FFA concentrations and increases with higher FFA levels [21]. Additionally, FFA activate protein kinase C (PKC) θ, a serine kinase that increases serine phosphorylation of IRS-1 [30].
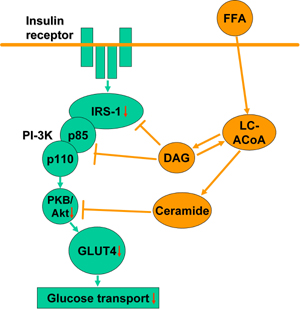 |
 |
Figure 1. FFA-induced impairment in insulin signaling within muscle cells. FFA enter muscle cells to form LC-ACoA, which are used in the synthesis of glycerolipids, such as DAG or TAG, or sphingolipids, such as ceramide. DAG and ceramide are able to inhibit insulin signal transduction within muscle cells. This causes decreased insulin stimulated GLUT4 translocation to the plasma membrane and, in consequence, decreased glucose transport to myocytes. FFA: free fatty acids. LC-ACoA: long-chain fatty acyl-CoA. IRS-1: insulin receptor substrate 1. PI-3K: phos-phatydiloinositol 3-kinase. PKB: protein kinase B. GLUT4: glucose transporter 4. |
|
It is generally accepted that FFA inhibit insulin signaling by modifying the intramyocellular lipid pool [13, 31]. Whole-body insulin sensitivity is negatively related to intramyocellular TAG content [4, 6]. Exercise training unexpectedly increased muscle TAG levels and improved insulin sensitivity [32]. There are also data suggesting that TAG accumulation protects against FFA-induced lipotoxicity [33]. Upregulation of diacylglycerol acyltransferase 1 (DGAT1), the enzyme that catalyzes the last step of TAG synthesis, prevents fat-induced insulin resistance [34, 35]. Therefore, it is assumed that muscle TAG does not exert unfavorable metabolic actions and might even be protective against accumulation of other, more toxic, lipid classes [31, 34, 35].
Diacylglycerol (DAG), a potent activator of PKC [36], may be an important lipid mediator of muscle insulin resistance. In healthy humans, 6h Intralipid/heparin infusion resulted in a fourfold increase in muscle DAG, together with an activation of PKC, its isoforms PKCβII and -δ as well as proinflammatory pathway IκB kinase/nuclear factor-κB [8]. The role of muscle DAG in the development of insulin resistance seems to be very important. However, there could be other lipid classes, i.e. sphingolipids, which might independently contribute to an impairment in insulin action.
Sphingomyelin signaling pathways
The pathways of sphingolipid metabolism are shown in Figure 2 [9, 12]. Ceramide is very active biologically and is involved in multiple cellular processes, including apoptosis and inflammation [9]. Ceramide might be generated during de novo synthesis from serine and palmitoyl-CoA and the first step in this process is catalyzed by the enzyme serine palmitoyltransferase (SPT) [37]. This enzyme is stimulated by saturated, but not unsaturated, fatty acids [38]. The key intermediate in de novo ceramide synthesis, which indicates the activity of the process, is sphinganine. At this step, a second fatty acyl-CoA, saturated or unsaturated, is added to sphinganine to form dihydroceramide.
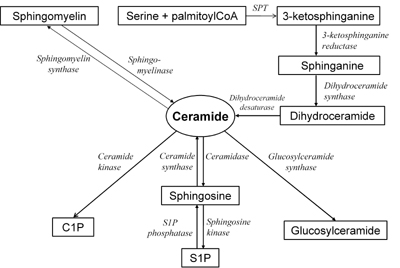 |
|
Figure 2. The pathways of sphingolipid metabolism. Ceramide, a second messenger in the sphngomyelin signaling pathway, is generated during hydrolysis of sphigomyelin or de novo synthesis from serine and palmitoyl-CoA. Ceramide might be deacylated to form sphingosine and fatty acid, phosphorylated to form C1P or glycosylated to form glucosylceramide. SPT: serine palmitoyltransferase. C1P: ceramide-1-phosphate. S1p: sphingosine-1-phos-phate. |
|
Ceramide is also generated through hydrolysis of plasma membrane sphingomyelin with the enzyme neutral sphingomyelinase or hydrolysis of lysosomal and endosomal sphingomyelin by acid sphingomyelinase [39]. Neutral sphingomyelinase is a target of tumor necrosis factor α (TNFα) action and is also activated by other inflammatory cytokines and steroids as well as by other stimuli [40, 41].
The deacylation of ceramide is catalyzed by the enzyme ceramidase. The products of this reaction are sphingosine and long-chain fatty acid [42]. Neutral and alkaline ceramidase are present in muscle, whereas acid ceramidase activity and mRNA are not found there [43]. Sphingosine is phosphorylated with sphingosine-1-kinase to form sphingosine-1-phosphate (S1P). Unlike ceramide, S1P was demonstrated to be involved in cellular proliferation, survival and protection against apoptosis [44].
Besides deacylation, ceramide might be converted back to sphingomyelin by the enzyme sphingomyelin synthase, which catalyzes the addition of phosphocholine as headgroup [45]. Another pathway of ceramide metabolism is phosphorylation by ceramide kinase. The product of this reaction is ceramide-1-phos-phate, which is supposed to be involved in eicosanoid biosynthesis [46]. Another pathway of ceramide metabolism is glycosylation with glucosylceramide synthase. Glucosylceramide, the compound synthesized in this reaction, is further modified in the Golgi apparatus to form complex glycosphingolipids, like gangliosides [47].
Mechanisms by which ceramide and other sphingolipids inhibit insulin action
Numerous in vitro studies indicate that ceramide may inhibit insulin signaling in muscle cells. The initially reported findings that ceramide inhibits insulin-stimulated tyrosine phosphorylation of IRS-1 [40] were not confirmed in further studies. The main mechanism of the impairment of insulin action in response to ceramide is a decrease in insulin-stimulated activation of PKB/Akt [48]. Ceramide blocks insulin stimulation of this kinase by two independent mechanisms, i.e. translocation of Akt to the plasma membrane and stimulation of Akt dephosphorylation by protein phosphatase 2A (PPA2) [49]. Blocking of Akt translocation to the plasma membrane requires atypical isoform PKC [50]. Inactivation of PKCζ negates the inhibitory effect of ceramide on Akt activation [51]. Similar effects were observed after inhibition of PPA2 in C2C12 myotubes [52]. A recent study provided evidence that an inhibition of insulin action by ceramide was partly independent of Akt and also involved PI-3K- and Rac-dependent remodeling of filamentous actin [53]. The consequence of the inhibitory action of ceramide on insulin signaling is decreased insulin-stimulated GLUT4 translocation to the plasma membrane [53, 54]. Additionally, ceramide-associated insulin resistance is directly linked to an enhanced apoptosis in skeletal muscle myotubes [55].
Ceramide might be a key factor in the insulin-desensitizing action of saturated fatty acids. Incubation of C2C12 myotubes with palmitate resulted in a twofold increase in ceramide content, with the concurrent inhibition of insulin-stimulated Akt phosphorylation. Similar effects were observed after incubation of myotubes with ceramide [48]. Additionally, it was demonstrated that blocking de novo ceramide synthesis by SPT inhibitors prevented an inactivation of Akt [52]. Overexepression of acid ceramidase also prevents an inhibitory effect of saturated fatty acids on insulin signaling [56]. Overexpression of carnitine palmitoyltransferase I, the rate-limiting enzyme in fatty acid import into mitochondria for oxidation, protected muscle cells from palmitate-induced insulin resistance and accumulation of DAG and ceramide [57]. The role that ceramide plays in mediating saturated fatty acid-induced insulin resistance was confirmed in the studies with cultured human myotubes [58, 59].
Ceramide is also supposed to mediate TNFα- and glucocorticoid-induced insulin resistance. As mentioned above, TNFα stimulated ceramide synthesis through activation of sphingomyelinases [40, 60]. The effects of adipocyte incubation with TNFα are mimicked by ceramides [61]. Furthermore, inhibition of ceramide synthesis prevents TNFα-induced insulin resistance [62]. Glucocorticoids increase membrane sphingomyelin content [63] and tissue ceramide through stimulation of both SPT and sphingomyelinases [64, 65].
It should be noted that the ceramide intermediate metabolites, sphinganine and sphingosine, also inhibit insulin-stimulated 2-deoxyglucose uptake in incubated rat soleus muscle [66]. The mechanism of this action remains unclear.
Another sphingolipid, which may be involved in the development of insulin resistance, is ganglioside GM3. This compound inhibits tyrosine phosphorylation of insulin receptors and IRS-1 [67]. It is possible that GM3 mediates TNFα-induced insulin resistance [67]. Inhibitors of glucosylceramide synthase prevent the effects of TNFα on insulin signaling [68] and enhance tyrosine phosphorylation of insulin receptors [69]. Improved phosphorylation of insulin receptors was also observed in GM3 synthase knockout mice [70].
Skeletal muscle ceramide and other sphingolipids in animal models of insulin resistance - the effect of insulin-sensitizing treatment
Turinsky et al. were the first to demonstrate that ceramide accumulates in the muscle of insulin-resistant animals in their study with Zucker fa/fa rats [71]. Muscle-specific overexpression of lipoprotein lipase resulted in muscle ceramide accumulation, together with the development of insulin resistance [14]. Increased muscle ceramide content was also reported in rats after high fat feeding [72], induction of streptozotocin diabetes [73] and treatment with dexamethazone [74]. In contrast, the data on the effect of lipid infusion on muscle ceramide content in rats are inconsistent, as both unchanged [74, 75] and increased ceramide content have been reported [76, 77]. In general, it is supposed that no increase in muscle ceramide levels under these conditions is related to the composition of lipid emulsion, which contains predominantly unsaturated fatty acids [10-12]. One hypothesis suggests that saturated fatty acids inhibit insulin signaling through an increase in muscle ceramide content, whereas unsaturated fatty acids might inhibit insulin signaling through an increase in DAG level [10-12]. The effect of lipid infusion on muscle ceramide will be discussed in more detail in the next section.
Decrease in muscle ceramide content is an effect of various strategies that improve insulin sensitivity. Knockout mutation of stearoyl-CoA desaturase 1 (SCD1) gene, results in both improvement in insulin sensitivity [78] and decrease in muscle ceramide content by inhibiting SPT and increasing β oxidation [79]. Although another study reported that SCD1 play a protective role against fatty acid-induced insulin resistance, an increase in muscle ceramide content also led to an inhibition of insulin signaling and glucose uptake [80]. Administration of ciliary neurotrophic factor prevents the development of lipid-induced insulin resistance and tissue ceramide accumulation [76].
The effect of thiazolidinediones on muscle ceramide level is less clear. It was demonstrated that pioglitazone and rosiglitazone prevented high fat feeding-induced insulin resistance and an increase in muscle ceramide [72, 81]. Interestingly, the study of Todd et al. found that ceramide was the only lipid class to be decreased after thiazolidinedione treatment and this decrease was accompanied by a reduction in inflammatory parameters in muscle [81]. In another study, 1-day treatment with troglitazone in mice resulted in decreased muscle ceramide content together with increased Akt protein expression [82]. In contrast, treatment of obese Zucker rats with rosiglitazone resulted in increased muscle ceramide content [83] and did not affect muscle ceramide level in high fat feeding rats [84]. In the latter paper, rosiglitazone also decreased insulin-stimulated glucose transport in skeletal muscle [84]. The discrepancies in results might come from the specific thiazolidenedione compound examined, the animal model used and the important differences in proportion between the periods of high fat feeding and thiazolidinedione treatment in the studies in question [72, 81, 84].
A single bout of exhaustive exercise leads to a decrease in muscle ceramide content in rats [85]. Interestingly, ceramide level after exercise is inversely related to 2-deoxyglucose uptake in each muscle type [85, 86]. In contrast, sphinganine and sphingosine were reported to be increased in muscle after acute exercise [87]. Exercise training consisting of 8 weeks of treadmill running in rats [88] or 1 week of swimming in mice [34] resulted in a decrease in muscle ceramide content. In the latter study, a decrease in muscle ceramide was associated with an increase in muscle DGAT1 activity and these results were replicated by transgenic overexpression of DGAT1 in muscle [34]. In contrast, another study reported no significant decrease in muscle ceramide level after exercise training in high fat fed rats [84].
Recently, an impressive study conducted by Holland et al. demonstrated beneficial effects of inhibiting ceramide synthesis in vivo [74]. Reducing tissue ceramide levels in rats with myriocin, an inhibitor of SPT, significantly negated dexamethazone-induced glucose intolerance, insulin resistance and inhibition of Akt in soleus muscle. Similarly, after genetic ablation of dihydroceramide desaturase 1, heterozygous mice demonstrated enhanced insulin sensitivity and were refractory to dexamethazone-induced insulin resistance. Myriocin treatment improved glucose tolerance and prevented the onset of diabetes in Zucker diabetic fatty rats, as well as reducing tissue ceramide content. Regarding the effect of myriocin in lipid-infused rats, the compound was able to prevent the development of insulin resistance after infusion of lard-oil/heparin, but not Intralipid. These results can be explained by the fact that lard-oil contains mostly saturated fatty acids, whereas Intralipid contains unsaturated fatty acids, which do not stimulate de novo ceramide synthesis [74].
In vivo inhibition of glycosphingolipid synthesis was reported in two studies using different animal models of insulin resistance [89, 90]. Treatment of Zucker diabetic fatty rats and diet-induced obese mice with glucosylceramide synthase inhibitor, Genz-123346, improved glucose tolerance and increased insulin signaling in muscle [89]. Another glucosylceramide synthase inhibitor, the iminosugar derivative AMP-DNM, reversed TNFα-induced abnormalities in insulin signal transduction in cultured 3T3-L1 adipocytes [90]. AMP-DNM also improved glucose tolerance and muscle and liver insulin sensitivity in vivo in ob/ob mice, high fat fed mice and Zucker diabetic fatty rats [90]. Treatment with both compounds was associated with marked reduction of glucosylceramide and ganglioside GM3 levels in muscle and liver [89, 90]. In contrast, ceramide content in liver [89, 90] and muscle [90] remained unchanged after treatment, indicating that ceramide is not the only sphingolipid involved in the pathogenesis of insulin resistance.
Ceramide in human skeletal muscle
In line with the results obtained in rodents, Adams et al. reported an almost twofold increase in muscle ceramide content in obese insulin-resistant humans [91]. In this study, negative correlation with baseline muscle ceramide level and insulin-stimulated Akt phosphorylation approached the level of significance [91]. We demonstrated a significant negative correlation between muscle ceramide content and insulin sensitivity in the group of apparently healthy males [20]. We also demonstrated a correlation between a decrease in insulin sensitivity and an increase in muscle ceramide during Intralipid infusion [20]. In another study, we observed an accumulation of muscle ceramide in subjects at risk of developing type 2 diabetes, i.e. in lean insulin-resistant offspring of type 2 diabetic subjects and in obese subjects with normal and impaired glucose tolerance [92]. Analysis of the activities of the enzymes regulating ceramide metabolism and of the content of ceramide intermediate metabolites suggested that in the offspring group this accumulation was related to decreased ceramide degradation, whereas in the obese subjects an increased de novo ceramide synthesis (in both groups) and sphingomyelin hydrolysis (in subjects with impaired glucose tolerance) were present [92].
In the study by Bruce et al., 8 weeks of exercise training in obese humans, resulted in a 42% decrease in muscle ceramide content, mostly attributable to a decrease in saturated ceramide species. The authors also observed a 15% decrease in muscle DAG level [93]. Similar effects of exercise training on muscle ceramide and DAG content in obese elderly population were observed by Dube et al. [94]. In this study, a significant correlation between a decrease in muscle ceramide and an improvement in insulin sensitivity during exercise training was also reported [94]. On the other hand, treatment of overweight nondiabetic subjects with conjugated linoleic acid (CLA), which resulted in a marked decrease in insulin sensitivity, as assessed on the basis of the oral glucose tolerance test, also led to an increase in muscle ceramide content, mostly through an increase in polyunsaturated ceramide species [95]. Positive correlation was noted between an increase in muscle ceramide level and an increase in the area under the curve for plasma glucose. Interestingly, muscle DAG and TAG remained unaffected by CLA treatment [95].
Other human studies were unable to demonstrate the potential role of muscle ceramide in modulating insulin action. In the study by Serlie et al., no difference in muscle ceramide content between lean and overweight and obese subjects was found [96]. Short term manipulation of plasma FFA did not affect muscle ceramide level [96]. Nor did treatment of obese type 2 diabetic subjects with pioglitazone decrease muscle ceramide level, despite improving insulin sensitivity [97]. No correlation between muscle ceramide content and insulin sensitivity was found after 38h [98] and 62h [99] periods of fasting. In the latter case, muscle ceramide tended to be higher after fasting. The authors observed a simultaneous decrease in Akt serine473 phosphorylation in response to insulin after fasting, but the relation of these changes was not reported [99].
Helge et al. observed no differences in muscle ceramide level between trained and untrained individuals, with the subsequent increase after a single bout of exercise in both groups [100]. In a recently published study, Skovbro et al. examined muscle ceramide content in four groups of subjects: healthy controls, endurance-trained persons and individuals with impaired glucose tolerance and type 2 diabetes [101]. Despite marked differences in insulin sensitivity, muscle ceramide was not different between the study groups, with the exception of higher values in trained subjects in comparison to those with impaired glucose tolerance [101]. The positive correlation between insulin sensitivity and muscle ceramide observed, however, lost its significance after trained individuals were excluded [101].
The differences in results between the studies are difficult to explain. In general, most studies have different purposes and analyze different aspects of the potential modulation of insulin action by ceramide. When the studies analyzing muscle ceramide content in subjects with different degrees of insulin sensitivity are considered, there are differences between our results [20, 92] and those obtained by Skovbro et al. [101].
Firstly, as has already been pointed out [102], there is a marked difference in muscle ceramide values between the studies (for instance for subjects with impaired glucose tolerance, approximately 900 nmol/g tissue in our study vs. approximately 150 nmol/g tissue in the study conducted by Skovbro et al.) [92, 101]. Both studies applied the same methodology for muscle ceramide measurements. The only difference is that we lyophilized muscle samples before analyses to avoid contamination with even small amounts of extracellular fat [20, 92]. However, when we compare our results with those obtained by Bruce et al. [93] and Thrush et al. [95], who used the same methodology but did not lyophilize muscle fibers, we found that the muscle ceramide values are relatively similar. For instance, in our obese subjects, muscle ceramide was 588 nmol/g tissue. Bruce et al. [93] obtained a value of 734 nmol/g tissue in the obese group in their study and the overweight/obese group in the study by Thrush et al. [95] yielded a value of 403 nmol/g tissue. Therefore, we do not think that muscle lyophilization could bias our results.
Secondly, there are differences in the relation between muscle ceramide and insulin sensitivity. The lack of increase in muscle ceramide levels in insulin-resistant individuals [101] contrasts with data obtained by Adams et al. [91]. Higher or unchanged muscle ceramide levels in endurance-trained individuals [101] are inconsistent with the results obtained in 2 studies, which demonstrated a decrease in muscle content of this compound after exercise training [93, 94]. The reason for these discrepancies remains unclear. All of the cited studies evaluated relatively small numbers of subjects. In such conditions, even slight differences in inclusion and exclusion criteria might influence the results. For instance, the majority of our subjects [92] was more than 25 years younger than those in the study of Skovbro et al. [101]. Other factors, such as dietary habits, smoking, etc. might also be significant. Additionally, no lean control group was analyzed in the study quoted [101]. As mentioned, both studies applied the same methodology [92, 101]. It should also be noted that this method does not differentiate between ceramide and biologically inactive dihydroceramide. Given the substantial differences in subjects' characteristics between the studies, one hypothesis is that in different groups of subjects the contribution of dihydroceramide to the total measurement may differ, and that this difference may influence the results.
Another issue is the potential effect of lipid infusion on muscle ceramide content. As mentioned above, it is assumed that infusion of Intralipid or Liposyn, which contain mainly unsaturated fatty acids, does not increase muscle ceramide [12]. Results supporting this assumption were obtained in rodents [74, 75] and humans [8, 96, 103]. Surprisingly, we observed an approximate 48% increase in muscle ceramide content after Intralipid/heparin infusion in healthy volunteers [20]. Although no comparable data have been obtained in humans, similar findings have been demonstrated in rats [76, 77]. There are two possible explanations for this phenomenon. Firstly, Intralipid does contain palmitate. Although the amount is as small as 8%, an increase plasma palmitate after Intralipid infusion has been reported (although in this study plasma palmitate was negatively related to muscle ceramide) [96]. Median plasma palmitate values after Intralipid infusion (239 μmol/l in the lean and 229 μmol/l in the obese group [96]) were only slightly lower than in another study (269 μmol/l total FFA level at the end of clamp [21]), where this level inhibited insulin signaling. Given the fact that total plasma FFA levels after Intralipid infusion were more that twofold higher in our study [20] in comparison to Serlie et al. [96], one hypothesis is that the plasma palmitate concentration was sufficient to stimulate de novo ceramide synthesis. Secondly, although unsaturated fatty acids do not stimulate ceramide de novo synthesis, they might enhance ceramide generation through an increase in sphingomyelinases activity [104]. DAG, which was demonstrated to increase after Intralipid infusion [8], was also able to increase acid sphingomyelinase activity [105]. It should also be noted that different results are available regarding the effect of lipid infusion on muscle DAG content, although DAG is generally believed to be a mediator of unsaturated fatty acid action. In contrast to the fourfold increase in muscle DAG content after Intralipid infusion reported by Itani et al. [8], no change in muscle DAG under such conditions was reported in another study [103].
Studies with negative results regarding an increase in muscle ceramide after Intralipid infusion indicate that this compound is not necessarily a lipid class that mediates the metabolic effects of unsaturated fatty acids. However, it is possible that under specific conditions (e.g. depending on the pre-study composition of plasma FFA, maximal plasma FFA concentrations during infusion or other factors), ceramide accumulation could accompany the phenomenon and aggravate the decrease in insulin action.
In summary, data on the role of muscle ceramide as a mediator of insulin resistance in humans remain inconsistent. Studies with negative results do not exclude a role for muscle ceramide in mediating insulin action, because of small sample sizes, methodological issues and because factors, such as ceramide bioactivity, its intracellular distribution or its rate of synthesis and degradation, were not measured. On the other hand, studies that reported significant correlations or differences between the groups do not prove the biological function of ceramide, because none of them demonstrated the cause-effect relationship. An analysis of all relevant studies indicates that the most probable explanation is that there are pathways of lipid-induced insulin resistance that do not involve muscle ceramide and that the role for ceramide as a mediator of muscle insulin resistance has its limitations. Ceramide should be viewed as one of multiple components in the complicated phenomenon of muscle insulin resistance. However, given the results of in vitro and animal data and the fact that four independent research groups have reported a relation between muscle ceramide and different aspects of insulin resistance in humans, it must be recognized that it is necessary to explore and determine the effects of this compound more closely in future studies.
To date, no data are available regarding skeletal muscle ganglioside content in relation to insulin resistance in humans. However, interesting results were reported by Langeveld et al. [106]. In this study, the authors examined 6 subjects with type I Gaucher disease, a lysosomal storage disorder, in which impaired breakdown of glucosylceramide leads to its accumulation in macrophages mainly situated in liver, spleen and bone marrow. It was demonstrated that subjects with Gaucher disease exhibit insulin resistance [106]. Plasma levels of glucosylceramide and ganglioside GM3 are increased in such individuals [107]. Whether Gaucher disease is associate with muscle glycosphingolipid accumulation, remains an open question.
Unresolved questions and directions for future research
As mentioned above, the final evidence for the role of muscle ceramide in modulating insulin action in humans is yet to be demonstrated. Such findings might have several important implications.
Firstly, metabolites of the sphingomyelin signaling pathway are also present in plasma. Sphingosine and sphinganine concentrations are increased in the plasma of type 2 diabetic subjects [108]. It would be interesting to examine whether some of the sphingolipids in plasma might serve as an early marker of insulin resistance.
Secondly, the question whether ceramide accumulation in muscle is a process secondary to an increased FFA delivery or a primary phenomenon, intrinsic for skeletal muscle, has not been extensively studied. Using cultured human muscle cells, it was demonstrated that the changes observed in myocellular fatty acids partitioning, i.e. decreased palmitate oxidation and increased esterification to phospholipids, might be of genetic origin [109]. Petersen et al. demonstrated an intramyocellular lipid accumulation and impairment in mitochondrial oxidative phosphorylation, despite normal local and systemic lipolysis in lean insulin-resistant offspring of type 2 diabetic subjects [110]. These findings cannot be applicable to ceramide, as no relationship between muscle ceramide and TAG was observed [92, 101] and no association with phospholipids has yet been established. Our studies with lean insulin-resistant offspring of type 2 diabetic subjects suggest that there could indeed be some intrinsic predisposition to muscle ceramide accumulation. These data, however, require confirmation in larger groups of subjects. Future studies could focus on analyzing the genes encoding the enzymes that regulate ceramide metabolism in the predisposition to type 2 diabetes or other conditions associated with insulin resistance.
Finally, the proof for the role of ceramide in the pathophysiology of human insulin resistance could be the demonstration that specific inhibition of ceramide synthesis leads to an improvement of insulin action and glucose tolerance in insulin-resistant or diabetic individuals.
References
- Ferrannini E. Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: problems and prospects. Endocr Rev 1998. 19:477-490. [DOD] [CrossRef]
- Fernandez-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr Rev 2003. 24:278-301. [DOD] [CrossRef]
- Vaag A, Henriksen JE, Beck-Nielsen H. Decrased insulin activation of glycogen synthase in skeletal muscles in young nonobese Caucasian first-degree relatives of patients with non-insulin-dependent diabetes mellitus. J Clin Invest 1992. 89:782-788. [DOD] [CrossRef]
- Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB, Storlien LH. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 1997. 46:983-988. [DOD] [CrossRef]
- Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W, Maerker E, Matthaei S, Schick F, Claussen CD, Haring HU. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 1999. 48:1113-1119. [DOD] [CrossRef]
- Perseghin G, Scifo P, De Cobelli F, Pagliato E, Battezzati A, Arcelloni C, Vanzulli A, Testolin G, Pozza G, Del Maschio A, Luzi L. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans. Diabetes 1999. 48:1600-1606. [DOD] [CrossRef]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 2004. 350:664-671. [DOD] [CrossRef]
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002. 51:2005-2011. [DOD] [CrossRef]
- Kolesnick R. The therapeutic potential of modulating the ceramide/ sphingomyelin pathway. J Clin Invest 2002. 110:3-8. [DOD]
- Summers SA, Nelson DH. A role for sphingolipids in producing the common features of type 2 diabetes, metabolic syndrome X, and Cushing's syndrome. Diabetes 2005. 54:591-602. [DOD] [CrossRef]
- Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res 2006. 45(1):42-72. [DOD] [CrossRef]
- Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev 2008. In press. [DOD]
- Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000. 106:171-176. [DOD] [CrossRef]
- Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, Lutz EP, Kako Y, Velez-Carrasco W, Goldberg IJ, Breslow JL, Shulman GI. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci USA 2001. 98:7522-7527. [DOD] [CrossRef]
- Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R, Abumrad NA. Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem 1999. 274:26761-26766. [DOD] [CrossRef]
- Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem 1999. 74:19055-19062. [DOD] [CrossRef]
- Kim JK, Gimeno RE, Higashimori T, Kim HJ, Choi H, Punreddy S, Mozell RL, Tan G, Stricker-Krongrad A, Hirsch DJ, et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest 2004. 113:756-763. [DOD]
- Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 1996. 97:2859-2865. [DOD] [CrossRef]
- Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, Olefsky JM. Fatty acid-induced insulin-resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. J Clin Endocrinol Metab 2002. 87:226-234. [DOD] [CrossRef]
- Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, Zendzian-Piotrowska M, Brzezinska Z, Gorski J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 2004. 53:1215-1221. [DOD] [CrossRef]
- Belfort R, Mandarino L, Kashyap S, Wirfel K, Pratipanawatr T, Berria R, DeFronzo RA, Cusi K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 2005. 54:1640-1648. [DOD] [CrossRef]
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose-fatty acid cycle; its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963. 1(7285):785-789. [DOD] [CrossRef]
- Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance. A reexamination. Diabetes 2000. 49:677-683. [DOD] [CrossRef]
- Mensink M, Blaak EE, van Baak MA, Wagnemakers AJM, Saris WHM. Plasma free fatty acid uptake and oxidation are already diminished in subjects at high risk for developing type 2 diabetes. Diabetes 2001. 50:2548-2554. [DOD] [CrossRef]
- Ruderman NB, Saha AK. Metabolic syndrome: adenosine monophosphate-activated protein kinase and malonyl coenzyme A. Obesity 2006. 14(Suppl 1):25S-33S. [DOD] [CrossRef]
- Roden M, Krssak M, Stingl H, Gruber S, Hofer A, Furnsinn C, Moser E, Waldhausl W. Rapid impairment of skeletal muscle transport/phosphorylation by free fatty acids in humans. Diabetes 1999. 48:358-364. [DOD] [CrossRef]
- Petersen KF, Hendler R, Price T, Perseghin G, Rothman DL, Held N, Amatruda JM, Shulman GI. 13C/31P-NMR studies on the mechanism of insulin resistance in obesity. Diabetes 1998. 47:381-386. [DOD] [CrossRef]
- Rothman DL, Magnusson I, Cline G, Gerard D, Kahn CR, Shulman RG, Shulman GI. Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci USA 1995. 92:983-987. [DOD] [CrossRef]
- Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidyloinositol 3-kinase activity. J Clin Invest 1999. 103:253-259. [DOD] [CrossRef]
- Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C θ and alterations in the insulin signaling cascade. Diabetes 1999. 48:1270-1274. [DOD] [CrossRef]
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003. 144:5159-5165. [DOD] [CrossRef]
- Hoppeler H, Howald H, Conley K, Lindstedt SL, Claasen H, Vock P, Weibel ER. Endurance training in humans: aerobic capacity and structure of skeletal muscle. J Appl Physiol 1985. 59:320-327. [DOD]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA 2003. 100(6):3077-3082. [DOD] [CrossRef]
- Liu L, Zhang Y, Chen N, Shi X, Tsang B, Yu YH. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest 2007. 117:1679-1689. [DOD] [CrossRef]
- Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest 2007. 117:1690-1698. [DOD] [CrossRef]
- Nishizuka Y. Protein kinase C and lipid signaling for sustained, cellular responses. Faseb J 1995. 9:484-496. [DOD]
- Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta 2003. 1632:16-30. [DOD]
- Merrill AH Jr. De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem 2002. 277:25843-25846. [DOD] [CrossRef]
- Kolesnick RN. 1,2-Diacylglycerols but not phorbol esters stimulate sphingomyelin hydrolysis in GH3 pituitary cells. J Biol Chem 1987. 262(35):16759-16762. [DOD]
- Kanety H, Hemi R, Papa MZ, Karasik A. Sphingomyelinase and ceramide suppress insulin-induced tyrosine phosphorylation of the insulin receptor substrate-1. J Biol Chem 1996. 271:9895-9897. [DOD] [CrossRef]
- Ramachandran CK, Murray DK, Nelson DH. Dexamethasone increases neutral sphingomyelinase activity and sphingosine levels in 3T3-L1 fibroblasts. Biochem Biophys Res Commun 1990. 167:607-613. [DOD] [CrossRef]
- Wertz PW, Downing DT. Ceramidase activity in porcine epidermis. FEBS Lett 1990. 268:110-112. [DOD] [CrossRef]
- Li CM, Hong SB, Kopal G, He X, Linke T, Hou WS, Koch J, Gatt S, Sandhoff K, Schuchman EH. Cloning and characterization of the full-length cDNA and genomic sequences encoding murine acid ceramidase. Genomics 1998. 50:267-274. [DOD] [CrossRef]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996. 381:800-803. [DOD] [CrossRef]
- Tafesse FG, Ternes P, Holthuis JC. The multigenic sphingomyelin synthase family. J Biol Chem 2006. 281:29421-29425. [DOD] [CrossRef]
- Lamour NF, Chalfant CE. Ceramide-1-phosphate: the "missing" link in eicosanoid biosynthesis and inflammation. Mol Interv 2005. 5:358-367. [DOD] [CrossRef]
- Yamashita T, Wu YP, Sandhoff R, Werth N, Mizukami H, Ellis JM, Dupree JL, Geyer R, Sandhoff K, Proia RL. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc Natl Acad Sci USA 2005. 102:2725-2730. [DOD] [CrossRef]
- Schmitz-Pfeiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 1999. 274:24202-24210. [DOD] [CrossRef]
- Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem 2004. 279:36608-36615. [DOD] [CrossRef]
- Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B(PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol 2003. 23(21):7794-7808. [DOD] [CrossRef]
- Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J 2004. 382:619-629. [DOD] [CrossRef]
- Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, Summers SA. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem 2003. 13:10297-10303. [DOD] [CrossRef]
- JeBailey L, Wanono O, Niu W, Roessler J, Rudich A, Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 2007. 56:394-403. [DOD] [CrossRef]
- Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol 1998. 18:5457-5464. [DOD]
- Turpin SM, Lancaster GI, Darby I, Febbraio MA, Watt MJ. Apoptosis in skeletal muscle myotubes is induced by ceramides and is positively related to insulin resistance. Am J Physiol 2006. 291(6):E1341-E1350. [DOD]
- Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem 2005. 280:20148-20153. [DOD] [CrossRef]
- Sebastian D, Herrero L, Serra D, Asins G, Hegardt FG. CPT I overexpression protects L6E9 muscle cells from fatty acid-induced insulin resistance. Am J Physiol 2007. 292:E677-E686. [DOD]
- Pickersgill L, Litherland GJ, Greenberg AS, Walker M, Yeaman SJ. Key role for ceramides in mediating insulin resistance in human muscle cells. J Biol Chem 2007. 282:12583-12589. [DOD] [CrossRef]
- Sabin MA, Stewart CE, Crowne EC, Turner SJ, Hunt LP, Welsh GI, Grohmann MJ, Holly JM, Shield JP. Fatty acid-induced defects in insulin signalling, in myotubes derived from children, are related to ceramide production from palmitate rather than the accumulation of intramyocellular lipid. J Cell Physiol 2007. 211:244-252. [DOD] [CrossRef]
- Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem 1996. 271:13018-13022. [DOD] [CrossRef]
- Wang CN, O'Brien L, Brindley DN. Effects of cell-permeable ceramides and tumor necrosis factor-alpha on insulin signaling and glucose uptake in 3T3-L1 adipocytes. Diabetes 1998. 47:24-31. [DOD] [CrossRef]
- Grigsby RJ, Dobrowsky RT. Inhibition of ceramide production reverses TNF-induced insulin resistance. Biochem Biophys Res Commun 2001. 287:1121-1124. [DOD] [CrossRef]
- Murray DK, Ruhmann-Wennhold A, Nelson DH. Dexamethasone effect on the phospholipid content of isolated fat cell ghosts from adrenalectomized rats. Endocrinology 1979. 105:774-777. [DOD]
- Linn SC, Kim HS, Keane EM, Andras LM, Wang E, Merrill AH Jr. Regulation of de novo sphingolipid biosynthesis and the toxic consequences of its disruption. Biochem Soc Trans 2002. 29:831-835. [DOD] [CrossRef]
- Lepine S, Lakatos B, Maziere P, Courageot MP, Sulpice JC, Giraud F. Involvement of sphingosine in dexamethasone-induced thymocyte apoptosis. Ann NY Acad Sci 2002. 973:190-193. [DOD]
- Turinsky J, Nagel GW. Effect of sphingoid bases on basal and insulin-stimulated 2-deoxyglucose transport in skeletal muscle. Biochem Biophys Res Commun 1992. 188:358-364. [DOD] [CrossRef]
- Kabayama K, Sato T, Kitamura F, Uemura S, Kang BW, Igarashi Y, Inokuchi J. TNFalpha-induced insulin resistance in adipocytes as a membrane microdomain disorder: involvement of ganglioside GM3. Glycobiology 2005. 15:21-29. [DOD] [CrossRef]
- Tagami S, Inokuchi Ji J, Kabayama K, Yoshimura H, Kitamura F, Uemura S, Ogawa C, Ishii A, Saito M, Ohtsuka Y, Sakaue S, Igarashi Y. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J Biol Chem 2002. 277(5):3085-3092. [DOD] [CrossRef]
- Yamashita T, Hashiramoto A, Haluzik M, Mizukami H, Beck S, Norton A, Kono M, Tsuji S, Daniotti JL, Werth N, Sandhoff R, Sandhoff K, Proia RL. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci USA 2003. 100:3445-3449. [DOD] [CrossRef]
- Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C, Komarnitsky S, Yew NS, Cheng SH. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 2007. 56:1210-1218. [DOD] [CrossRef]
- Turinsky J, O'Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem 1990. 265:16880-16885. [DOD]
- Zendzian-Piotrowska M, Baranowski M, Zabielski P, Gorski J. Effects of pioglitazone and high-fat diet on ceramide metabolism in rat skeletal muscles. J Physiol Pharmacol 2006. 57(Suppl 10):101-114. [DOD]
- Gorska M, Dobrzyn A, Zendzian-Piotrowska M, Gorski J. Effect of streptozotocin-diabetes on the functioning of the sphingomyelin-signalling pathway in skeletal muscles of the rat. Horm Metab Res 2004. 36:14-21. [DOD] [CrossRef]
- Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 2007. 5:167-179. [DOD] [CrossRef]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 2002. 277:50230-50236. [DOD] [CrossRef]
- Watt MJ, Hevener A, Lancaster GI, Febbraio MA. Ciliary neurotrophic factor prevents acute lipid-induced insulin resistance by attenuating ceramide accumulation and phosphorylation of c-Jun N-terminal kinase in peripheral tissues. Endocrinology 2006. 147:2077-2085. [DOD] [CrossRef]
- Dube JJ, Bhatt BA, Dedousis N, Bonen A, O'Doherty RM. Leptin, skeletal muscle lipids, and lipid-induced insulin resistance. Am J Physiol 2007. 293:R642-R650. [DOD]
- Rahman SM, Dobrzyn A, Dobrzyn P, Lee SH, Miyazaki M, Ntambi JM. Stearoyl-CoA desaturase 1 deficiency elevates insulin-signaling components and down-regulates protein-tyrosine phosphatase 1B in muscle. Proc Natl Acad Sci USA 2003. 100:11110-11115. [DOD] [CrossRef]
- Dobrzyn A, Dobrzyn P, Lee SH, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, Friedman JM, Ntambi JM. Stearoyl-CoA desaturase-1 deficiency reduces ceramide synthesis by downregulating serine palmitoyltransferase and increasing beta-oxidation in skeletal muscle. Am J Physiol 2005. 288:E599-E607. [DOD]
- Pinnamanemi SK, Southgate RJ, Febbraio MA, Watt MJ. Stearoyl CoA desaturase 1 is elevated in obesity but protects against fatty acid-induced skeletal muscle insulin resistance in vitro. Diabetologia 2006. 49:3027-3037. [DOD] [CrossRef]
- Todd MK, Watt MJ, Le J, Hevener AL, Turcotte LP. Thiazolidinediones enhance skeletal muscle triacylglycerol synthesis while protecting against fatty acid-induced inflammation and insulin resistance. Am J Physiol 2007. 292:E485-E493. [DOD]
- Planavila A, Alegret M, Sanchez RM, Rodriguez-Calvo R, Laguna JC, Vazquez-Carrera M. Increased Akt protein expression is associated with decreased ceramide content in skeletal muscle of troglitazone-treated mice. Biochem Pharmacol 2005. 69:1195-1204. [DOD] [CrossRef]
- Lessard SJ, Lo Giudice SL, Lau W, Reid JJ, Turner N, Febbraio MA, Hawley JA, Watt MJ. Rosiglitazone enhances glucose tolerance by mechanisms other than reduction of fatty acid accumulation within skeletal muscle. Endocrinology 2004. 145:5665-5670. [DOD] [CrossRef]
- Lessard SJ, Rivas DA, Chen ZP, Bonen A, Febbraio MA, Reeder DW, Kemp BE, Yaspelkis BB 3rd, Hawley JA. Tissue-specific effects of rosiglitazone and exercise in the treatment of lipid-induced insulin resistance. Diabetes 2007. 56:1856-1864. [DOD] [CrossRef]
- Dobrzyn A, Gorski J. Ceramides and sphingomyelins in skeletal muscles of the rat: content and composition. Effect of prolonged exercise. Am J Physiol 2002. 282:E277-E285. [DOD]
- Gorski J, Dobrzyn A, Zendzian-Piotrowska M. The sphingomyelin-signaling pathway in skeletal muscles and its role in regulation of glucose uptake. Ann NY Acad Sci 2002. 967:236-248. [DOD]
- Dobrzyn A, Gorski J. Effect of acute exercise on the content of free sphinganine and sphingosine in different skeletal muscle types of the rat. Horm Metab Res 2002. 34:523-529. [DOD] [CrossRef]
- Smith AC, Mullen KL, Junkin KA, Nickerson J, Chabowski A, Bonen A, Dyck DJ. Metformin and exercise reduce muscle FAT/CD36 and lipid accumulation and blunt the progression of high-fat diet-induced hyperglycemia. Am J Physiol 2007. 293:E172-E181. [DOD] [CrossRef]
- Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C, Komarnitsky S, Yew NS, Cheng SH. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 2007. 56:1210-1218. [DOD] [CrossRef]
- Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, Aten J, Kuipers F, Serlie MJ, Wennekes T, et al. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 2007. 56:1341-1349. [DOD] [CrossRef]
- Adams JM 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004. 53:25-31. [DOD] [CrossRef]
- Straczkowski M, Kowalska I, Baranowski M, Nikolajuk A, Otziomek E, Zabielski P, Adamska A, Blachnio A, Gorski J, Gorska M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 2007. 50:2366-2373. [DOD] [CrossRef]
- Bruce CR, Thrush AB, Mertz VA, Bezaire V, Chabowski A, Heigenhauser GJ, Dyck DJ. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am J Physiol 2006. 291:E99-E107. [DOD]
- Dube JJ, Amati F, Stefanovic-Racic M, Toledo FG, Sauers SE, Goodpaster BH. Exercise-induced alterations in intramyocellular lipids and insulin resistance: the athlete’s paradox revisited. Am J Physiol 2008. 294:E882-E888. [DOD]
- Thrush AB, Chabowski A, Heigenhauser GJ, McBride BW, Or-Rashid M, Dyck DJ. Conjugated linoleic acid increases skeletal muscle ceramide content and decreases insulin sensitivity in overweight, non-diabetic humans. Appl Physiol Nutr Metab 2007. 32:372-382. [DOD] [CrossRef]
- Serlie MJ, Meijer AJ, Groener JE, Duran M, Endert E, Fliers E, Aerts JM, Sauerwein HP. Short-term manipulation of plasma free fatty acids does not change skeletal muscle concentrations of ceramide and glucosylceramide in lean and overweight subjects. J Clin Endocrinol Metab 2007. 92:1524-1529. [DOD] [CrossRef]
- Serlie MJ, Allick G, Groener JE, Ackermans MT, Heijligenberg R, Voermans BC, Aerts JM, Meijer AJ, Sauerwein HP. Chronic treatment with pioglitazone does not protect obese patients with diabetes mellitus type II from free fatty acid-induced insulin resistance. J Clin Endocrinol Metab 2007. 92:166-171. [DOD] [CrossRef]
- Soeters MR, Sauerwein HP, Groener JE, Aerts JM, Ackermans MT, Glatz JF, Fliers E, Serlie MJ. Gender-related differences in the metabolic response to fasting. J Clin Endocrinol Metab 2007. 92:3646-3652. [DOD] [CrossRef]
- Soeters MR, Sauerwein HP, Dubbelhuis PF, Groener JE, Ackermans MT, Fliers E, Aerts JM, Serlie MJ. Muscle adaptation to short-term fasting in healthy lean humans. J Clin Endocrinol Metab 2008. In press. [DOD]
- Helge JW, Dobrzyn A, Saltin B, Gorski J. Exercise and training effects on ceramide metabolism in human skeletal muscle. Exp Physiol 2004. 89:119-127. [DOD] [CrossRef]
- Skovbro M, Baranowski M, Skov-Jensen C, Flint A, Dela F, Gorski J, Helge JW. Human skeletal muscle ceramide content is not a major factor in muscle insulin sensitivity. Diabetologia 2008. 51(7):1253-1260. [DOD]
- Boden G. Ceramide: a contributor to insulin resistance or an innocent bystander? Diabetologia 2008. 51(7):1095-1096. [DOD]
- Vistisen B, Hellgren LI, Vadset T, Scheede-Bergdahl C, Helge JW, Dela F, Stallknecht B. Effect of gender on lipid-induced insulin resistance in obese subjects. Eur J Endocrinol 2008. 158:61-68. [DOD] [CrossRef]
- Worgall TS, Johnson RA, Seo T, Gierens H, Deckelbaum RJ. Unsaturated fatty acid-mediated decreases in sterol regulatory element-mediated gene transcription are linked to cellular sphingolipid metabolism. J Biol Chem 2002. 277:3878-3885. [DOD] [CrossRef]
- Kolesnick RN. 1,2-Diacylglycerols but not phorbol esters stimulate sphingomyelin hydrolysis in GH3 pituitary cells. J Biol Chem 1987. 262:16759-16762. [DOD]
- Langeveld M, Ghauharali KJ, Sauerwein HP, Ackermans MT, Groener JE, Hollak CE, Aerts JM, Serlie MJ. Type I Gaucher disease, a glycosphingolipid storage disorder, is associated with insulin resistance. J Clin Endocrinol Metab 2008. 93:845-851. [DOD] [CrossRef]
- Ghauharali-van der Vlugt K, Langeveld M, Poppema A, Kuiper S, Hollak CE, Aerts JM, Groener JE. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin Chim Acta 2008. 389:109-113. [DOD]
- Gorska M, Dobrzyn A, Baranowski M. Concentrations of sphingosine and sphinganine in plasma of patients with type 2 diabetes. Med Sci Monit 2005. 11:CR35-CR38. [DOD]
- Gaster M, Rustan AC, Aas V, Beck-Nielsen H. Reduced lipid oxidation in skeletal muscle from type 2 diabetic subjects may be of genetic origin: evidence from cultured myotubes. Diabetes 2004. 53:542-548. [DOD] [CrossRef]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 2004. 350:664-671. [DOD] [CrossRef]
This article has been cited by other articles:

|
Ceramide-mediated insulin resistance and impairment of cognitive-motor functions
de la Monte SM, Tong M, Nguyen V, Setshedi M, Longato L, Wands JR
J Alzheimers Dis 2010. 21(3):967-984
|
|

|
Sphingolipids and insulin resistance: the five Ws
Summers SA
Curr Opin Lipidol 2010. 21(2):128-135
|
|

|
Associations of energy intake and type 2 diabetes with hypertryglyceridemia in older adults living in the Mediterranean Islands: The Medis Study
Tyrovolas S, Pounis G, Zeimbekis A, Antonopoulou M, Bountziouka V, Gotsis E, Metallinos G, Polystipioti A, Polychronopoulos E, Lionis C, Panagiotakos DB
J Nutr Elder 2010. 29(1):72-86
|
|

|
Skeletal muscle insulin resistance in endocrine disease
Peppa M, Koliaki C, Nikolopoulos P, Raptis SA
J Biomed Biotechnol 2010. 2010:527850
|
|

|
Ceramides: A new player in the inflammation-insulin resistance paradigm?
Gill JM, Sattar N
Diabetologia 2009. 52(12):2475-2477
|
|

|
Metabolite profiling identifies candidate markers reflecting the clinical adaptations associated with Roux-en-Y gastric bypass surgery
Mutch DM, Fuhrmann JC, Rein D, Wiemer JC, Bouillot JL, Poitou C, Clement K
PLoS One 2009. 4(11):e7905
|
|

|
Insulin resistance and neurodegeneration: roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis
de la Monte SM, Longato L, Tong M, Wands JR
Curr Opin Investig Drugs 2009. 10(10):1049-1060
|
|

|
Factors Associated with the Prevalence of Diabetes Mellitus Among Elderly Men and Women Living in Mediterranean Islands: The MEDIS Study
Tyrovolas S, Zeimbekis A, Bountziouka V, Voutsa K, Pounis G, Papoutsou S, Metallinos G, Ladoukaki E, Polychronopoulos E, Lionis C, Panagiotakos DB
Rev Diabet Stud 2009. 6(1):54-63
|
|
|


)
)

