Original Data
| Rev Diabet Stud,
2008,
5(3):144-153 |
DOI 10.1900/RDS.2008.5.144 |
Incomplete Killing And Enhanced Activation of Islet-Reactive CD8+ T Cells by FasL-Expressing Dendritic Cells Limits Protection from Diabetes
Mikael Maksimow1,2, Catharina Alam1, Arno Hänninen1
1Department of Medical Microbiology and Immunology, and MediCity Research Laboratory, University of Turku, Finland
2Turku Graduate School of Biomedical Sciences, Turku, Finland
Address correspondence to: Arno Hänninen, e-mail: arno.hanninen@utu.fi
Manuscript submitted September 23, 2008; resubmitted November 11, 2008; accepted November 26, 2008.
Keywords: type 1 diabetes, autoimmunity, dendritic cell, FasL, CD8, islet-antigen, tolerance, vaccination
Abstract
AIMS: Autologous dendritic cells (DC) are a promising tool for induction of cytotoxic CD8+ T cell immunity against tumors and chronic viral infections. When armed with the death-inducing Fas-ligand (FasL, CD195), DC attenuate delayed-type hypersensitivity reactions and allotransplant rejection by promoting activation-induced cell death in T cells. We investigated the possibility of using FasL-expressing DC to induce deletion of islet-reactive CD8+ T cells in vivo, and to prevent destruction of pancreatic islets in a model of autoimmune diabetes. METHODS: DC, propagated from mouse bone marrow cells, were purified and made to express FasL and islet-antigen via plasmid transfection. CD8+ T cells (OT-I cells) recognizing the antigen, ovalbumin, were adoptively transferred to transgenic mice expressing ovalbumin in islets (RIP-OVAlo mice), and these mice were primed with ovalbumin. To test the potential of DC to prevent diabetes in this model, the mice were later intravenously vaccinated with the transfected DC. RESULTS: Transfected DC induced partial deletion of antigen-reactive CD8+ T cells in vivo and reduced the level of lymphocyte infiltration into pancreatic islets. Diabetes developed less frequently in vaccinated mice, but this effect was limited. Further in vitro analysis showed that FasL-expressing DC not only deleted many of the responding CD8+ T cells but also promoted the expansion of surviving cells and their IFN-γ production. CONCLUSIONS: FasL-expressing DC can also have stimulatory effects on CD8+ T cells warranting further investigation into the optimal design of tolerance-promoting DC-vaccination to prevent autoimmune diabetes.
Introduction
Adaptive immune responses are initiated in lymph nodes by an interaction between dendritic cells (DC) and T cells displaying suitable antigen specificity towards a peptide presented on the surface of the DC [1]. Activation of a T cell depends on different factors such as strength and duration of signals received via its T cell, cytokine and costimulatory receptors [2-4], strength of cell-to-cell adhesion with DC [5], and availability of essential nutritional factors such as tryptophan [6]. Shortly after their activation, T cells also become susceptible to signaling via death-inducing receptors, which can lead to programmed cell death (PCD) and apoptosis [7, 8]. The death receptor Fas (CD95) belongs to the tumor necrosis factor (TNF) receptor family and induces apoptosis upon ligand binding via activation of the intracellular caspase cascade [9-11]. Activated T cells express Fas early in their life cycle and begin to co-express FasL at a later stage, leading to autocrine and paracrine apoptosis signaling known as activation-induced cell death (AICD) [12].
Interestingly, deletion (via PCD) of ovalbumin-reactive CD8+ T cells, after activation in pancreatic (and kidney-draining) lymph nodes of RIP-mOVA mice, was previously shown to occur equally in wild-type or TNFR2-deficient cells, but not in cells from a lpr/lpr (Fas-deficient) background [13]. Thus, signaling via Fas appears to be critical for physical elimination of islet-reactive CD8+ T cells and maintenance of self-tolerance in this model of autoimmunity. Moreover, defects in Fas-FasL signaling are associated with lymphoproliferation and increased production of autoantibodies [14], and the Fas/FasL pathway has also been implicated in the pathogenesis of several autoimmune diseases [15-19].
Matsue and coworkers employed genetic modification of DC by plasmid transfection to produce DC expressing FasL. When pulsed with antigen, these DC suppress in vivo delayed-type hypersensitivity reactions and contact hypersensitivity reactions [20]. When given together with an allogenic bone marrow transplantation, FasL-expressing DC of the same allotype prevent a graft-versus host response [21]. As several lines of evidence indicate an important role for CD8+ T cells in autoimmune diabetes [22-25], we decided to investigate the effects of a DC vaccination consisting of FasL-expressing DC on the development of diabetes during ongoing beta-cell destruction by islet-specific CD8+ T cells. We used the RIP-OVAlo transgenic model expressing ovalbumin in islets [26], and immunized them with ovalbumin to ensure maximal activation of adoptively transferred, ovalbumin-reactive CD8+ T cells. Our results indicated that vaccination with DC co-expressing ovalbumin and FasL reduced the number of ovalbumin-reactive CD8+ T cells in pancreatic lymph nodes and spleen, and formation of inflammatory infiltrates (“insulitis”) in islets. However, preservation of islet-function achieved by vaccination with FasL and ovalbumin co-expressing DC was limited. In vitro analyses indicated enhanced activity of surviving T cells, possibly explaining the limited effect on islet destruction in the in vivo situation.
Materials and methods
Mice and cell lines
OT-I mice [26], carrying transgenic T cell receptors (TCR) specific for MHC class I restricted chicken ovalbumin (OVA) peptides, were used as donors for naïve T cells. RIP-OVAlo mice expressing OVA under the control of a rat insulin promoter [26] were used as recipients of these cells. Both these transgenic mouse strains were originally generated by Drs. L.C. Heath, J. Allison and F.R. Carbone in the Walter and Eliza Hall Institute, Melbourne, Australia.
Rag2-/- mice were used as donors of bone-marrow cells for DC propagation. All the above-mentioned mice had the same C57BL/6 background. Mice were bred and maintained under specific pathogen-free (SPF) conditions in Central Animal Laboratory in Turku University and all animal experiments were approved by the Institutional Ethical Committee of Turku University.
Plasmid construction
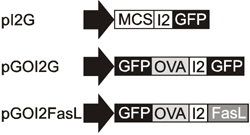
In this study, DC were engineered to express cell surface Fas ligand together with a model autoantigen (OVA), in an attempt to induce deletion of autoantigen-specific T cells after DC vaccination. To ensure that all of the nucleofected DC would express both the antigen and the death-receptor ligand (FasL), a bicistronic expression vector was used. The pIRES2EGFP vector (Clontech; Palo Alto, CA) was chosen and modified by introducing a silent mutation to one MscI site located in the kanamycin/neomycin resistance gene (resulting in a plasmid named pI2G), thus allowing the replacement of the EGFP gene using the other MscI site just upstream of the gene. A cDNA encoding the GFP-OVA fusion protein was digested from the pGO plasmid [27] using NheI and EcoRI restriction enzymes to the multiple cloning site of pI2G to produce the pGOI2G plasmid. A plasmid encoding both the GFP-OVA fusion protein and the death ligand FasL was constructed by replacing the EGFP gene downstream of the IRES2 sequence with FasL cDNA using MscI and NotI restriction enzymes. The Fas ligand was cloned from activated OT-I T cells using gene specific primers: N-terminal primer 5’ GATTACTGGCCACAACC ATG CAG CAG CCC ATG AAT TAC CC and C-terminal primer 5’ CGATATGCGGCCGCT TTA AAG CTT ATA CAA GCC G (MscI and NotI sites are underlined and FasL binding sequences are in italics). The purified PCR fragment was digested with MscI and NotI restriction enzymes and then ligated with similarly digested pGOI2G plasmid to produce the pGOI2FasL plasmid. Gene constructs are depicted in Figure 1.
 |
 |
Figure 1. Gene constructs used to express the autoantigen and FasL. Plasmid pGOI2G encoding the OVA antigen as a GFP fusion protein was constructed using the pI2G (pIRES2EGFP) backbone. Plasmid encoding for both the GFP-OVA and death ligand FasL was generated by replacing the GFP gene downstream of IRES (I2) sequence with FasL cDNA to produce the plasmid pGOI2FasL. |
|
Nucleofection of dendritic cells
DC were propagated from bone-marrow cells according to a modified method designed by Inaba et al. [28, 29]. After 5 days of culture the DC were activated using 10 μg/ml of anti-CD40 antibody (clone HM40-3, Pharmingen). After overnight culture, DC were purified using anti-CD11c-MACS-beads and MACS-LSTM columns (Miltenyi Biotech, Bergisch Gladbach, Germany). Purified CD11c+ DC cells were counted and used in nucleofection. The Amaxa nucleofection system was used, as previously, optimized for mouse BM-DCs [27]. Briefly, 24 hours after activation the cells were counted and up to 5 × 106 cells were suspended in the "5034" nucleofection solution, mixed with 5 μg of plasmid DNA and immediately nucleofected using the T-24 program. After nucleofection, the cells were cultured overnight in complete medium (RPMI 1640 supplemented with 10% FCS, 20 mM L-glutamine, 5 × 10-5 M 2-mercaptoethanol, penicillin/streptomycin), in the presence of 20 μg/ml recombinant murine GM-CSF (Immunotools, Friesoythe, Germany), in 5 ml polypropylene tubes (Becton Dickinson, San Jose, CA) at a density of 1 × 106 cells/ml/tube.
The next day, the expression level of activation markers on nucleofected DC was analyzed by flow cytometry. The cells were collected, washed twice and labeled with phycoerythin (PE) conjugated anti-CD86 or anti-CD40, using anti-rat IgG2a as a control antibody (Ab). MHC class II (IAb) and FasL were detected using specific primary Abs (KH74 and Kay-10, respectively) and PE-conjugated anti-mouse IgG secondary Ab. Anti-human VAP-1 Abs TK-8-14 and 2D10, prepared in our laboratory, were used as species- and isotype-matched control Abs in these stainings. All other antibodies were from Pharmingen/Becton and Dickinson. The cells were analyzed using FACScalibur flow cytometer (Becton and Dickinson) and the expression levels were analyzed from GFP+ cells using WinMDI 2.8 software (http://facs.scripps.edu/software.html).
T cell preparation and activation
Naïve OT-I T cells were prepared from the lymph nodes and spleens of OT-I mice using standard techniques. Their numbers were determined by labeling the cells with FITC-conjugated anti-CD8 (Pharmingen) and the H2-Kb-SIINFEKL-specific tetramer (Beckman Coulter, Fullerton, CA). This tetramer consists of the MHC-restriction element and the particular 8-mer linear peptide sequence (SIINFEKL). When these 2 elements are combined they are recognized by the TCR of OT-I cells. The purified naïve OT-I cells were then used either for adoptive cell transfer or for in vitro experiments as responder T cells in co-cultures with nucleofected DC, in complete medium supplemented with 50 μM 2-mercaptoethanol.
CFSE dilution assays were used to analyze T cell activation by nucleofected DC. After nucleofection, DC were rested overnight (o/n) and then the cells were collected, washed and counted. Subsequently, 5000 viable DC were plated on a flat-bottom 96-well plate together with 0.5 × 106 CFSE labeled OT-I cells. On day 3 and 5, the cells were collected and the CFSE dilution in viable (7-AAD-) cells was analyzed using flow cytometry.
T cell activation was also analyzed by measuring IFN-γ production from culture supernatants. For this purpose, viable nucleofected DC and naïve T cells were plated at the DC-T cell ratio of 1:200, 1:80 and 1:40 in a round-bottom 96-well plate (0.2 × 106 T cells). The culture supernatants were collected at day 3. IFN-γ levels were measured using specific ELISA with anti-IFN-γ (R4 6A2) as capture antibody and with biotinylated anti-IFN-γ (XMG1.2) as the detecting antibody (both from Pharmingen/Becton and Dickinson).
Induction of autoimmune diabetes and vaccination with DC
To study the effect of DC vaccination on autoimmune diabetes, RIP-OVAlo mice were adoptively transferred with 2.5 × 106 OT-I T cells (cell number determined as explained above) and immunized subcutaneously with 200 μg of ovalbumin (Grade V, Sigma) in incomplete Freund’s adjuvant, to allow effective activation of OT-I T cells and reliable disease induction [30, 31]. On day 5 thereafter, mice were vaccinated intravenously using 1 × 106 DC nucleofected one hour earlier. Blood glucose levels were measured on day 10 and 14 using MediSenseTM Glucometer. Mice with blood glucose levels greater than 14.3 mmol/l at one or two readings were considered to be diabetic. To study the effect of DC vaccination on lymphocyte infiltration into pancreatic islets, RIP-OVAlo mice were sacrificed on day 7 or day 11 after disease induction and DC vaccination, as explained above. The level of lymphocyte infiltration (insulitis) was evaluated, as described below.
To examine the effect of DC vaccination on antigen-specific T cell number in vivo, a larger number (4 × 106) of OT-I T cells were transferred to allow their sufficient recovery for physical detection. In these experiments, recipient RIP-OVAlo mice were killed 2 days after the DC vaccination, and lymphocytes were isolated from the spleen and pancreas-draining lymph nodes. The percentage of OVA-specific CD8+ T cells was measured using flow cytometry after isolated cell suspensions were labeled with anti-CD8 and H-2Kb-SIINFEKL-specific tetramers.
Determination of insulitis
Insulitis scoring was determined using hematoxylin-eosin stain. Stained sections of paraffin-embedded pancreata (50-100 islets from different parts of each pancreas) were evaluated for the level of lymphocytic infiltration into each islet. Each islet was given a score (0-3) depending on whether it was free of infiltrating cells (0), had minor or sparse infiltration in islet periphery (1), was more densely infiltrated but had more than half of islet area intact (2), or was densely infiltrated into more than half of the total islet area (3). From the total analyzed islets, the mean score was calculated, representing the insulitis score. Each pancreas was assessed in a blinded fashion by one investigator. Photomicrographs were taken with Olympus BX51 microscope using DP Manager software.
Statistical methods
Statistical significance for differences in the numbers of OT-I cells in mice vaccinated with one of the three different DC preparations was determined using a one-way ANOVA with Bonferroni's correction. Significance of differences in the level of insulitis between the two groups was determined by a nonparametric Mann-Whitney test. Differences in the proportion of mice developing diabetes were evaluated using the Fisher’s exact test.
Results
Activated DC can be engineered to co-express antigen and Fas ligand
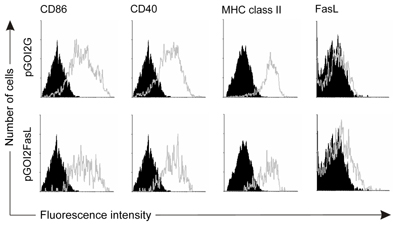
When DC were nucleofected with pGOI2G, all of the successfully nucleofected DC (i.e. GFP+ cells, typically 80% of DC which remained viable o/n) were highly positive for CD86 (B7.2), CD40 and MHC class II, but did not express detectable levels of FasL (Figure 2, upper row). When DC were nucleofected with pGOI2FasL, all the GFP+ cells expressed equal levels of activation markers and FasL up to a detectable level (Figure 2, lower row). This was comparable to previous reports following FasL transfection [21].
 |
|
Figure 2. Nucleofected DC have the phenotype of activated DC and can be engineered to express FasL. DC were nucleofected with pGOI2G (upper row), or pGOI2FasL plasmid (lower row) and the next day CD86, CD40, MHC class II and FasL expression was assessed by flow cytometry. The specific staining of nucleofected (GFP+) DC is shown as light grey open histograms and control staining is shown as black histograms. |
|
DC-expressing antigen and Fas ligand are able to induce partial deletion of islet-specific CD8+ T cells in vivo
In order to justify the use of nucleofected DC as in vivo modulators of CD8+ T cell response, it was necessary to ensure that after they were adoptively transferred to recipient animals, they were able to migrate to lymphoid tissues, surviving sufficiently to be able to encounter and activate antigen-specific T cells. In these experiments, we used conventional C57BL/6 mice, not receiving any antigen immunization but only nucleofected DC. Therefore, we were able to use the in vivo proliferation of OT-I T cells as a read-out of DC "grafting".
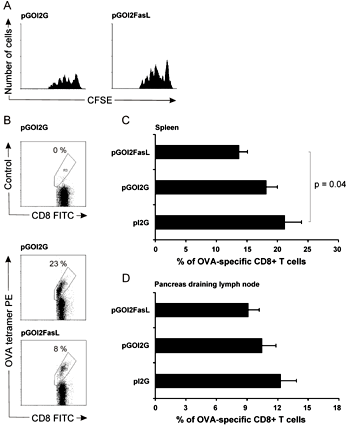
CFSE-labeled OT-I T cells, analyzed 3 days after their transfer, proliferated in the spleen (Figure 3A) and lymph nodes (not shown) of mice which had received 1 × 106 nucleofected DC one hour preceding OT-I transfer. Proliferation in mice transferred with antigen-only was comparable to mice transferred with FasL co-expressing DC, indicating that FasL co-expressing DC achieved comparable grafting to recipient mice. To test for their potency to specifically kill activated CD8+ T cells involved in autoimmune tissue destruction, 1 × 106 nucleofected DC were injected i.v. into RIP-OVAlo mice, transferred with OT-I T cells 5 days earlier and were primed with OVA to induce T cell activation. Two days after i.v. injection of DC, the percentage of OT-I T cells among total CD8+ T cells in the spleen and in the pancreas draining lymph nodes was calculated from the analysis.
Mice receiving the DC vaccine (pI2G) had approximately 21% of their CD8+ T cells reactive with the OVA-specific tetramer (a representative staining is shown in Figure 3B). On average, mice receiving DC expressing OVA and FasL had 14% tetramer-reactive CD8+ T cells, and thus FasL co-expression resulted in a 33% reduction in the relative number of OT-I T cells in the spleen (Figure 3C). This reduction was also visible in the pancreas-draining lymph nodes, but the effect did not achieve statistical significance (Figure 3D). Mice receiving DC nucleofected with pGOI2G also had slightly less OVA-specific T cells compared to control-vaccinated mice, suggesting a low level of cell-death induction by mere antigen re-encounter on DC at a vulnerable stage.
 |
|
Figure 3. DC expressing OVA and FasL are able to become grafted in recipient mice and can reduce the numbers of activated OVA-specific CD8+ T cells in lymphoid tissues. A: Proliferation of CFSE-labeled (naïve) OT-I cells at day 3 can be detected in the spleen of mice receiving an i.v. injection of either antigen-only expressing DC (left), or DC co-expressing FasL (right), and is comparable between the two cell populations. B: A representative staining of OT-I cells (“OVA-tetramer PE”) from a mouse that received DC nucleofected either with pGOI2G or pGOI2FasL, i.e. with antigen only or FasL co-expressing DC. C: On average, the reduction in OVA-specific CD8 T cells was from 21.3% to 13.8% in the spleen (p = 0.04), and also reduced slightly (from 12.3% to 9%, not significant) in pancreas-draining lymph nodes (D). Percentages of OT-I cells are expressed as cells reactive with H-2Kb-SIINFEKL-specific tetramer among CD8+ T cells as determined by flow cytometry in mice vaccinated with 1 × 106 DC nucleofected with indicated plasmids. In panels C and D, results are mean ± SD of three individual experiments in which mice were vaccinated with pI2G (7 mice), pGOI2G (9 mice) and pGOI2FasL (9 mice). |
|
Infiltration of lymphocytes into pancreatic islet is reduced in mice vaccinated with DC expressing OVA and Fas ligand
As DC expressing both the antigen and FasL were able to induce partial in vivo deletion of islet-specific CD8+ T cells in the OT-I T cell population, the effect of the vaccine on lymphocyte infiltration into pancreatic islets of RIP-OVAlo mice was investigated. Diabetes was induced in RIP-OVAlo mice as detailed in the methods section. Nucleofected DC (1 × 106 DC) were injected in the mice on day 5, and mice were sacrificed on day 7 or day 11 and analyzed for lymphocyte infiltration into pancreatic islets.
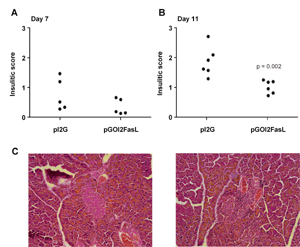
As changes in the level of insulitis are a relatively insensitive measure of treatment efficacy, we chose to vaccinate the control group with DC expressing the pI2G plasmid (i.e. with no antigen) instead of DC expressing the pGOI2G plasmid (i.e. with antigen). Since DC expressing the pGOI2G plasmid (i.e. antigen) resulted in a slight reduction in the numbers of OT-I cells in the spleen (as depicted in Figure 3C) it was regarded to be less suitable as a control vaccine when determining the natural level of insulitis in this model. In the group receiving DC expressing the pI2G plasmid, the level of insulitis was anticipated to fall into the natural range. On day 7 mice vaccinated with control DC (pI2G) displayed slightly more insulitis compared to mice vaccinated with DC nucleofected with pGOI2FasL (Figure 4A), and this difference became statistically significant on day 11 (Figure 4B).
 |
|
Figure 4. Mice vaccinated with DC expressing OVA and FasL (pGOI2FasL) have less islet-infiltrating lymphocytes compared to mice vaccinated with control DC (pI2G). RIP-OVAlo mice received the DC vaccination 5 days after disease induction and on day 7 (A) or day 11 (B), lymphocyte infiltration into the pancreatic islets was analyzed. None of the mice in A were included in the analyses presented in Figure 3. C: A representative islet without insulitis (left panel) and with insulitis (right panel) from an animal treated with either pGOI2FasL- or pI2G-nucleofected DC, respectively. |
|
Vaccination with FasL co-expressing DC protects a limited number of mice from diabetes
As vaccination with DC expressing antigen and FasL (from the pGOI2FasL plasmid) was able to both reduce the numbers of OT-I T cells in the spleen (by roughly one third) and decrease lymphocyte infiltration into pancreatic islets, we investigated if this treatment would also protect mice from diabetes. Therefore, RIP-OVAlo mice were observed for the occurrence of hyperglycemia, indicating immune-mediated islet-cell destruction or dysfunction. To study if FasL co-expressing DC reduced diabetes specifically due to FasL and antigen, we chose to vaccinate the control group with DC expressing antigen (not an "empty" plasmid).
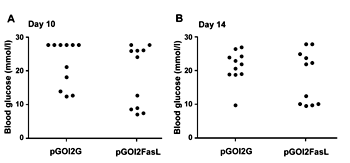
Blood glucose levels were measured on day 10 and 14 after the mice were sacrificed. On day 10, 8 and 6 mice out of 11, respectively, vaccinated with either OVA encoding or OVA + FasL encoding DC had become diabetic (Figure 5A; p = 0.36 using Fisher's exact test). By day 14, two more mice in the control group had become diabetic (p = 0.15 using Fisher's exact test) (Figure 5B).
 |
|
Figure 5. FasL co-expressing DC protect only a limited number of mice against islet-destruction by CD8+ T cells. RIP-OVAlo mice, induced to develop autoimmune diabetes, were vaccinated on day 5 using DC nucleofected with the indicated plasmids. Blood glucose levels were measured on day 10 (A) and 14 (B). Mice with blood glucose levels greater than 14.3 mmol/l were considered to be diabetic. |
|
FasL co-expressing DC induce CD8+ T cell responses stronger than normal DC
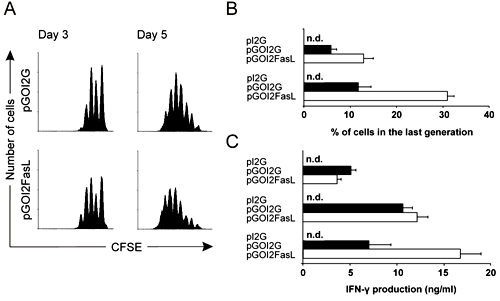
As injection of FasL co-expressing DC was able to cause partial deletion of OT-I T cells in vivo, yet incomplete alleviation of islet destruction, we examined the effects of these DC in an in vitro system. The response of naïve and pre-activated CD8+ T cells was measured using the same OT-I T cells as in the in vivo deletion experiments. The proliferation of naïve OT-I T cells induced by nucleofected DC required correct antigen presentation because control DC (pI2G) did not induce any proliferation. DC expressing OVA and FasL (pGOI2FasL) induced more vigorous cell division than DC expressing the OVA antigen only (pGOI2G) (Figure 6A). The percentage of cells dividing 3 or more times was calculated from the CFSE peaks. Results obtained from this analysis indicated an acceleration of OT-1 T-cell cell division (Figure 6B). At the highest DC-T cell ratio (1:40), DC expressing OVA and FasL induced more than a twofold increase in IFN-γ production in naïve OT-I cells (Figure 6C).
 |
|
Figure 6. DC nucleofected with pGOI2FasL (i.e. Fas co-expressing DC) induce proliferation and IFN-γ production of naïve CD8+ T cells. After nucleofection the DC were rested o/n and then used to activate OT-I T cells. A: Proliferation of naïve OT-I T cells was determined using the CFSE dilution assay on day 3 (left panels) and day 5 (right panels) in the presence of DC nucleofected with pGOI2G (top panels) and pGOI2FasL (bottom panels). Only viable (7-AAD-) cells are shown. B: From the CFSE peaks, the percentage of cells dividing 3 or more times were counted on day 3 (top) and day 5 (bottom). C: IFN-γ production of naïve OT-I T cells activated by 1000 (top), 2500 (middle) or 5000 (bottom) viable nucleofected DC was measured from the culture supernatant on day 3 using specific ELISA. (n.d. = not detectable). The CFSE histograms are representative of three experiments and the IFN-γ and proliferation data are shown as the average of 3 to 4 experiments ± SEM. |
|
Discussion
In this study, the effects of DC expressing both an islet-antigen and the death-ligand FasL on CD8+ T cell activation were investigated. We tested if such DC were able to attenuate islet destruction mediated by islet-reactive CD8+ T cells in vivo. We chose to express both the antigen and Fas ligand from a single bicistronic expression cassette, to ensure that none of the nucleofected DC would express antigen without the death ligand. Nucleofection of DC with the pGOI2FasL plasmid or the pGOI2G control plasmid resulted in a population of highly activated DC, and nucleofection with pGOI2FasL plasmid also enabled expression of FasL. Using such DC as a vaccine, we found it was possible to reduce both the numbers of antigen-specific CD8+ T cells in animals and the level of insulitis in their pancreatic islets, but the effect on the development of diabetes was more limited. We chose to inject 1 × 106 nucleofected DC because we were able to detect proliferation of naïve OT-I cells in response to this number of injected (OVA-expressing) DC, and because this allowed a reasonable number of mice per experiment to be treated. Although in this setting, the difference in diabetes incidence did not achieve statistical significance. The number of mice remaining normoglycemic was greater in the group receiving FasL co-expressing DC. It remains to be determined if vaccinating a greater number of injected DC or a greater number of mice with these DC would have allowed the detection of a beneficial effect in the development of diabetes.
Our results showed that FasL-expressing DC can become grafted in lymphoid tissues of recipient mice. Although they were able to reduce the number of pre-activated CD8+ T cells specific for an antigen co-expressed by the DC, FasL-expressing DC enhanced proliferation of surviving CD8+ T cells and their IFN-γ production in vitro. More than a decade ago Alderson and coworkers found that Fas-ligation using an agonistic antibody enhanced proliferation of anti-CD3-stimulated human peripheral blood mononuclear cells in vitro [32]. Later, this was confirmed using recombinant FasL-antibody chimeras for Fas-ligation, and shown to involve caspase activation [33]. Our group also studied this phenomenon earlier, and extended it to activation of antigen-specific CD4+ T cells by natural antigen-presenting cells, namely DC [34]. Moreover, absence of functional Fas on diabetogenic CD4+ T cells (BDC2.5 T cells) has been shown to dampen their pathogenic potential due to a defect in their proliferation and IL-2 production [35]. Thus, the concept of Fas-mediated costimulation of T cells and its relevance for in vivo T cell-mediated response is strongly supported, and, according to the present study, could be extended to involve antigen-specific CD8+ T cells.
When we applied FasL co-expressing DC in vivo, they were able to significantly reduce the numbers of ovalbumin-specific CD8+ T cells in the spleen and, to a lesser extent, also in pancreatic lymph nodes. This could be a result of better targeting of intravenously injected DC into the spleen compared to the pancreatic lymph node. Vaccination with these DC also decreased the level of insulitis in the RIP-OVA mice expressing ovalbumin as a model autoantigen in islets. However, development of hyperglycemia in the majority of vaccinated mice, together with a decrease in the level of cellular infiltrates in pancreatic islets suggests that effector CD8+ T cells surviving Fas-engagement by FasL co-expressing DC possessed stronger effector functions on a per cell basis. Because islet beta-cells are susceptible to various inflammatory mediators including IFN-γ [36, 37], this paradox could be explained by the finding that FasL-costimulated OT-I cells produced elevated amounts of IFN-γ. In addition, numerous target cells can be killed by a single CD8+ cytotoxic T cell before its own demise [38], suggesting that if hyperstimulated, fewer cytotoxic T cells could account for a similar level of target cell destruction via cellular cytotoxicity.
In some earlier studies, where DC engineered to co-express FasL have been applied to induce tolerance, they have been ineffective or shown adverse effects, such as pulmonary inflammation and pleuritis [39, 40]. Although we observed no such adverse side effects in our mice, our present study is reminiscent of work where beneficial effects were either incomplete or accompanied by adverse effects. In studies by Matsue et al. [20, 21] and Min et al. [41], FasL co-expressing DC prevented delayed-type hypersensitivity reactions, allograft rejection and graft-versus-host reactions successfully.
At the first glance, these different outcomes may look conflicting. However, the biology of DC is complicated and possible differences between the DC used by different investigators must not be overlooked. In studies by Matsue et al. [20, 21] for instance, DC were transfected with FasL using particle-mediated gene delivery device. Differences in the transfection methodology, the plasmid used, and the nature, source and characteristics of DC could all account for the differences observed in the function of FasL co-expressing DC. Moreover, factors related to differences in target cells and tissues, and in priming and activation of effector CD4+ and/or CD8+ T cells could also explain the variability in the final effects of FasL co-expressing DC in different models of immune-mediated diseases. Thus, it seems reasonable to conclude that FasL co-expressing DC, while representing a potentially effective and promising form of antigen-specific immunosuppression, need to be evaluated carefully in yet other relevant models of autoimmune diabetes before their translation to clinical trials with patients exhibiting risk of developing type 1 diabetes.
Acknowledgments:
We thank Anne Sovikoski-Georgieva for secretarial assistance, Anne Rumpunen-Virtanen and Hanna Suodenjärvi for breeding and maintenance of the mouse colonies, and Minna Santanen for technical assistance. The study was supported by the Finnish Diabetes Research Foundation, Finnish Academy, Sigrid Juselius Foundation (Finland), Juvenile Diabetes Research Foundation International, Päivikki and Sakari Sohlberg Foundation, and Turku Graduate School of Biomedical Sciences.
References
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998. 392:245-252. [DOD] [CrossRef]
- Lanzavecchia A, Sallusto F. Antigen decoding by T lymphocytes: from synapses to fate determination. Nat Immunol 2001. 2:487-492. [DOD] [CrossRef]
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996. 14:233-258. [DOD] [CrossRef]
- Schluns KS, Lefrancois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol 2003. 3:269-279. [DOD] [CrossRef]
- Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol 2007. 7:131-143. [DOD] [CrossRef]
- Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 2004. 4:762-774. [DOD] [CrossRef]
- Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science 1998. 280:243-248. [DOD] [CrossRef]
- Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol 2006. 6:196-204. [DOD] [CrossRef]
- Gravestein LA, Borst J. Tumor necrosis factor receptor family members in the immune system. Semin Immunol 1998. 10:423-434. [DOD] [CrossRef]
- Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem 2000. 69:217-245. [DOD] [CrossRef]
- Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 2004. 21:461-465. [DOD]
- Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev 2003. 193:70-81. [DOD] [CrossRef]
- Kurts C, Heath WR, Kosaka H, Miller JF, Carbone FR. The peripheral deletion of autoreactive CD8+ T cells induced by cross-presentation of self-antigens involves signaling through CD95 (Fas, Apo-1). J Exp Med 1998. 188:415-420. [DOD] [CrossRef]
- Kimura M, Matsuzawa A. Autoimmunity in mice bearing lprcg: a novel mutant gene. Int Rev Immunol 1994. 11:193-210. [DOD] [CrossRef]
- Suvannavejh GC, Dal Canto MC, Matis LA, Miller SD. Fas-mediated apoptosis in clinical remissions of relapsing experimental autoimmune encephalomyelitis. J Clin Invest 2000. 105:223-231. [DOD] [CrossRef]
- Sabelko-Downes KA, Gimenez MT, Suvannavejh GC, Miller SD, Russell JH. Genetic control of pathogenic mechanisms in autoimmune demyelinating disease. J Neuroimmunol 2000. 110:168-176. [DOD] [CrossRef]
- Stassi G, Di Liberto D, Todaro M, Zeuner A, Ricci-Vitiani L, Stoppacciaro A, Ruco L, Farina F, Zummo G, De Maria R. Control of target cell survival in thyroid autoimmunity by T helper cytokines via regulation of apoptotic proteins. Nat Immunol 2000. 1:483-488. [DOD] [CrossRef]
- Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, et al. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med 1997. 186:613-618. [DOD] [CrossRef]
- Xue C, Lan-Lan W, Bei C, Jie C, Wei-Hua F. Abnormal Fas/FasL and caspase-3-mediated apoptotic signaling pathways of T lymphocyte subset in patients with systemic lupus erythematosus. Cell Immunol 2006. 239:121-128. [DOD] [CrossRef]
- Matsue H, Matsue K, Walters M, Okumura K, Yagita H, Takashima A. Induction of antigen-specific immunosuppression by CD95L cDNA-transfected 'killer' dendritic cells. Nat Med 1999. 5:930-937. [DOD] [CrossRef]
- Matsue H, Matsue K, Kusuhara M, Kumamoto T, Okumura K, Yagita H, Takashima A. Immunosuppressive properties of CD95L-transduced "killer" hybrids created by fusing donor- and recipient-derived dendritic cells. Blood 2001. 98:3465-3472. [DOD] [CrossRef]
- Wong FS, Visintin I, Wen L, Flavell RA, Janeway CA Jr. CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J Exp Med 1996. 183:67-76. [DOD] [CrossRef]
- Wicker LS, Leiter EH, Todd JA, Renjilian RJ, Peterson E, Fischer PA, Podolin PL, Zijlstra M, Jaenisch R, Peterson LB. Beta 2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes 1994. 43:500-504. [DOD] [CrossRef]
- Serreze DV, Chapman HD, Varnum DS, Gerling I, Leiter EH, Shultz LD. Initiation of autoimmune diabetes in NOD/Lt mice is MHC class I-dependent. J Immunol 1997. 158:3978-3986. [DOD]
- Kagi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice. J Exp Med 1997. 186:989-997. [DOD] [CrossRef]
- Miller JF, Kurts C, Allison J, Kosaka H, Carbone F, Heath WR. Induction of peripheral CD8+ T-cell tolerance by cross-presentation of self antigens. Immunol Rev 1998. 165:267-277. [DOD] [CrossRef]
- Maksimow M, Miiluniemi M, Marttila-Ichihara F, Jalkanen S, Hanninen A. Antigen targeting to endosomal pathway in dendritic cell vaccination activates regulatory T cells and attenuates tumor immunity. Blood 2006. 108:1298-1305. [DOD] [CrossRef]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med 1992. 176:1693-1702. [DOD] [CrossRef]
- Maksimow M, Santanen M, Jalkanen S, Hanninen A. Responding naive T cells differ in their sensitivity to Fas engagement: early death of many T cells is compensated by costimulation of surviving T cells. Blood 2003. 101:4022-4028. [DOD] [CrossRef]
- Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med 1996. 184:923-930. [DOD] [CrossRef]
- Hanninen A, Nurmela R, Maksimow M, Heino J, Jalkanen S, Kurts C. Islet beta-cell-specific T cells can use different homing mechanisms to infiltrate and destroy pancreatic islets. Am J Pathol 2007. 170:240-250. [DOD] [CrossRef]
- Alderson MR, Armitage RJ, Maraskovsky E, Tough TW, Roux E, Schooley K, Ramsdell F, Lynch DH. Fas transduces activation signals in normal human T lymphocytes. J Exp Med 1993. 178:2231-2235. [DOD] [CrossRef]
- Kennedy DO, Kojima A, Yano Y, Hasuma T, Otani S, Matsui-Yuasa I. Growth inhibitory effect of green tea extract in Ehrlich ascites tumor cells involves cytochrome c release and caspase activation. Cancer Lett 2001. 166:9-15. [DOD] [CrossRef]
- Maksimow M, Soderstrom TS, Jalkanen S, Eriksson JE, Hanninen A. Fas costimulation of naive CD4 T cells is controlled by NF-kappaB signaling and caspase activity. J Leukoc Biol 2006. 79:369-377. [DOD] [CrossRef]
- Vence L, Benoist C, Mathis D. Fas deficiency prevents type 1 diabetes by inducing hyporesponsiveness in islet beta-cell-reactive T-cells. Diabetes 2004. 53:2797-2803. [DOD] [CrossRef]
- Rabinovitch A. An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Diabetes Metab Rev 1998. 14:129-151. [DOD] [CrossRef]
- Wachlin G, Augstein P, Schroder D, Kuttler B, Kloting I, Heinke P, Schmidt S. IL-1beta, IFN-gamma and TNF-alpha increase vulnerability of pancreatic beta cells to autoimmune destruction. J Autoimmun 2003. 20:303-312. [DOD] [CrossRef]
- Bossi G, Trambas C, Booth S, Clark R, Stinchcombe J, Griffiths GM. The secretory synapse: the secrets of a serial killer. Immunol Rev 2002. 189:152-160. [DOD] [CrossRef]
- Buonocore S, Van Meirvenne S, Demoor FX, Paulart F, Thielemans K, Goldman M, Flamand V. Dendritic cells transduced with viral interleukin 10 or Fas ligand: no evidence for induction of allotolerance in vivo. Transplantation 2002. 73:S27-S30. [DOD] [CrossRef]
- Buonocore S, Flamand V, Claessen N, Heeringa P, Goldman M, Florquin S. Dendritic cells overexpressing Fas-ligand induce pulmonary vasculitis in mice. Clin Exp Immunol 2004. 137:74-80. [DOD] [CrossRef]
- Min WP, Gorczynski R, Huang XY, Kushida M, Kim P, Obataki M, Lei J, Suri RM, Cattral MS. Dendritic cells genetically engineered to express Fas ligand induce donor-specific hyporesponsiveness and prolong allograft survival. J Immunol 2000. 164:161-167. [DOD]
|


)
)
)
)
)
)

