Review
| Rev Diabet Stud,
2009,
6(3):187-202 |
DOI 10.1900/RDS.2009.6.187 |
The Beneficial Effects of C-Peptide on Diabetic Polyneuropathy
Hideki Kamiya1,2, Weixian Zhang1, Anders A.F. Sima1,3
1Department of Pathology, Wayne State University, Detroit, MI, USA
2Department of Endocrinology and Diabetes, Nagoya University, Nagoya, Japan
3Department of Neurology, Wayne State University, Detroit, MI, USA
Address correspondence to: Anders A.F. Sima, e-mail: asima@med.wayne.edu
Manuscript submitted October 3, 2009; resubmitted October 15, 2009; accepted October 25, 2009.
Keywords: type 1 diabetes, C-peptide, diabetic neuropathy, BB/Wor-rat, inflammation, hyperlipidemia, nerve growth factor, neuroregulation, neurotrophic factors
Abstract
Diabetic polyneuropathy (DPN) is a common complication in diabetes. At present, there is no adequate treatment, and DPN is often debilitating for patients. It is a heterogeneous disorder and differs in type 1 and type 2 diabetes. An important underlying factor in type 1 DPN is insulin deficiency. Proinsulin C-peptide is a critical element in the cascade of events. In this review, we describe the physiological role of C-peptide and how it provides an insulin-like signaling function. Such effects translate into beneficial outcomes in early metabolic perturbations of neural Na+/K+-ATPase and nitric oxide (NO) with subsequent preventive effects on early nerve dysfunction. Further corrective consequences resulting from this signaling cascade have beneficial effects on gene regulation of early gene responses, neurotrophic factors, their receptors, and the insulin receptor itself. This may lead to preventive and corrective results to nerve fiber degeneration and loss, as well as, promotion of nerve fiber regeneration with respect to sensory somatic fibers and small nociceptive nerve fibers. A characteristic abnormality of type 1 DPN is nodal and paranodal degeneration with severe consequences for myelinated fiber function. This review deals in detail with the underlying insulin-deficiency-related molecular changes and their correction by C-peptide. Based on these observations, it is evident that continuous maintenance of insulin-like actions by C-peptide is needed in peripheral nerve to minimize the sequences of metabolic and molecular abnormalities, thereby ameliorating neuropathic complications. There is now ample evidence demonstrating that C-peptide replacement in type 1 diabetes promotes insulin action and signaling activities in a more enhanced, prolonged, and continuous fashion than does insulin alone. It is therefore necessary to replace C-peptide to physiological levels in diabetic patients. This will have substantial beneficial effects on type 1 DPN.
Abbreviations: Aβ - sensory nerve fiber, type 2; ADP - adenosine diphosphate; AMP - adenosine monophosphate; ankyrinG - cell membrane adaptor protein; Bax - Bcl-2–associated X protein (promotes apoptosis); BB/Wor rat - BioBreeding/Worcester rat (animal model of spontaneous autoimmune diabetes); BBZDR/Wor - BioBreeding Zucker Diabetic Rat/Worcester (animal model of spontaneously non-insulin-dependent diabetes); Bcl-2 - B-cell lymphoma 2; Ca2+- bivalent ionic calcium; Caspr - contactin-associated protein; CGRP - calcitonin gene-related peptide; CNS - central nervous system; C-peptide - connecting peptide; CREB - cyclic AMP response element-binding protein; Cdk5- cyclin-dependent kinase 5; DCCT - Diabetes Control and Complications Trial; DPN - diabetic polyneuropathy; DRG - dorsal root ganglion; ERK - extracellular signal-regulated kinase; GSK-3β - glycogen synthase kinase 3 beta; HbA1c - glycated hemoglobin; HSP27 - heat shock protein 27 (inhibitor of apoptosis, regulator of cell development); HSP70 - heat shock protein 70 (inhibitor of apoptosis, regulator of cell development); IGF - insulin-like growth factor; IR - insulin receptor; JNK - c-Jun N-terminal kinase; MAP kinase - mitogen-activated protein kinase; mRNA - messenger ribonucleic acid; Na+ - sodium ion; NADPH - nicotinamide adenine dinucleotide phosphate; Na+/K+-ATPase - sodium, potassium adenosintriphosphatase, also sodium-potassium pump; NCV - nerve conduction velocity; NGF - nerve growth factor ; NF - neurofilament; NFH - neurofilament protein, heavy chain; NFL - neurofilament protein, light chain; NFM - neurofilament medium polypeptide; NO - nitric oxide; eNOS - endothelial nitric oxide synthase; NF-κB - nuclear factor-kappa light-chain enhancer of activated B cells; NT-3 - neurotrophin-3; O-GlcNAc - O-glycoside-linked N-acetylglucosamine; PARP - poly ADP-ribose polymerase-1; PI-3-kinase - phosphatidylinositide-3-kinase; RPTP-β - receptor-like protein tyrosine phosphatase beta; SAPK - stress-activated protein kinase; SH3 domain - SRC homology 3 domain; SOD - superoxide dismutase; STZ - streptozotocin; Trk - tropomyosin receptor kinase (also known as tyrosine kinase); TrkA - tyrosine kinase A (highly affine to the NGF receptor); Zn2+ - bivalent ionic zinc
Introduction
Diabetic polyneuropathy (DPN) is the most common late complication of diabetes, with serious clinical consequences such as pain, sensory loss, foot ulceration, and potential amputation of limbs. DPN involves somatic, nociceptive, and autonomic peripheral nerves. The latter can have severe consequences for cardiac and gastrointestinal functions [1]. DPN affects both type 1 and type 2 diabetes patients, although specific differences exist in the underlying pathobiology, pathology, and clinical expression of the disease [2]. The progression and the expression of the disease is sometimes more rapid and severe in type 1 DPN [3, 4].
In recent past decades, several attempts have been made to address the underlying mechanisms therapeutically, e.g. through inhibiting an activated polyol pathway, oxidative stress, or by replacement of acetylcarnitine. However, the clinical benefits remain limited [5-8]. In retrospect, these failures were probably, at least in part, due to interventions coming too late in the natural history of the disease. DPN is a highly dynamic disorder with changing underlying pathobiological mechanisms that require early intervention. Another likely reason for limited success in treating this disorder, is the misconception that hyperglycemia is the only underlying cause of DPN. This assumption led to the premise that DPN complicating type 1 and type 2 diabetes is the same [1, 4].
Whilst hyperglycemia is significant in the development of DPN, the DCCT and other trials have shown that hyperglycemic control has only moderate effects [9-11]. Other correctable underlying pathogenetic factors may be equally important. These other factors may differ according to the type of diabetes. In type 1 diabetes, insulin deficiency per se plays a prominent role, due to the many functions of insulin other than controlling hyperglycemia [12, 13]. With respect to the nervous system, insulin is probably the most potent nerve growth factor, through gene regulatory control of other growth factors, their receptors, neuroregulation, and modulation of innate inflammatory processes [14-19].
In type 2 diabetes, the opposite problem occurs, namely hyperinsulinemia, which is implicated in several underlying mechanisms. A second component in type 2 DPN, not commonly considered, is accompanying hyperlipidemia. In a recent large scale study, we demonstrated that elevated triglyceride levels were significantly correlated with rapid progression of DPN [20].
Coming back to type 1 diabetes, it is known that repeated insulin injections, although controlling hyperglycemia, are not sufficient to correct the various cellular and regulatory functions of insulin. As insulin is secreted from the pancreatic β-cells, C-peptide is secreted in equimolar quantities and has a substantially longer half-life [21]. It has been suggested that the insulin-like effects of C-peptide are mediated via its interaction with insulin. Natural insulin exists in a hexameric form kept together by zinc-ions [22, 23]. As shown in detail in an accompanying paper in this volume [24], C-peptide appears to regulate the dehexamirization of insulin by binding to Zn2+. It thereby enhances and prolongs the many direct nonhypoglycemic effects of insulin. This shows that C-peptide has an important interactive regulatory role on insulin, which would explain the insulinomimetic effects of C-peptide [25, 26].
In this review, we discuss the effects of C-peptide on several mechanisms underlying DPN and the resulting clinical effects. For the last several decades we have made use of a rat model of type 1 diabetes, with spontaneous onset of diabetes consequent to an immune-mediated β-cell destruction [27], that mimics type 1 diabetes in humans closely. This BB/Wor-rat model is totally insulin- and C-peptide-deficient and requires daily small insulin doses for survival (Table 1). In this review article, we occasionally contrast this model with the spontaneously hyperinsulinemic type 2 diabetic BBZDR/Wor-rat. The latter shows the same degree of hyperglycemia, onset of diabetes at the same age, and outbred on the same genetic background as the type 1 BB/Wor-rat [28]. Finally, we describe the beneficial effects of C-peptide when administered to patients with type 1 DPN.
Table
1.
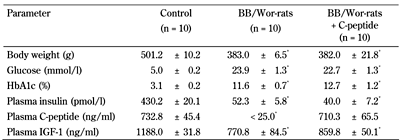
Metabolic profiles of control, diabetic, and C-peptide-treated rats |
|
 |
 |
Legend:
The metabolic status of eight-month diabetic BB/Wor-rats treated with small maintenance doses (1.6-2.6 U/day) of insulin. This treatment is compared with that of BB/Wor-rats receiving full replacement doses (75nM/kg/d) of rat II C-peptide alone delivered subcutaneously via osmopumps from onset of diabetes. These data contrast to those of age-matched non-diabetic control rats. Note, C-peptide has no direct effect on glucose levels. * p < 0.001 vs. control rats. |
|
The physiological role of C-peptide in the peripheral nerve
Following its discovery by Steiner et al. [29, 30], it was believed that C-peptide would have an insulin-like glucose lowering effect. Since this could not be substantiated, C-peptide was dismissed as a non-functional peptide. Also, in the type 1 diabetic BB/Wor-rat, C-peptide restitution does not influence blood glucose or HbA1c levels (Table 1).
In the 1990's, the Karolinska group, and subsequently others, demonstrated effects on blood-flow, early diabetic neuropathy and nephropathy in type 1 diabetes patients [31-33], which led to renewed interest in C-peptide physiology. It was shown that C-peptide binds specifically to cell surfaces, and it was suggested that C-peptide acted via a G-protein-related receptor mechanism [34, 35]. Detailed studies by Grunberger et al. demonstrated that C-peptide autophosphorylates the insulin receptor in the presence of insulin [25, 36, 37]. Also, it stimulates p38 MAP-kinase and PI-3 kinase activities, and diminishes the activation of JNK phosphorylation with subsequent dose-related effects on Na+/K+-ATPase activity and nitric oxide (NO) [38-40]. Further downstream effects related to C-peptide include anti-apoptotic effects and gene-regulatory effects on neurotrophic factors, via several transcription factors, such as c-jun, c-fos and NF-κB [41-44]. NF-κB also plays a crucial role in the anti-inflammatory effects of C-peptide [18, 19].
However, despite years of effort, the attempts to identify a specific C-peptide receptor have failed. Instead, recent studies have revealed interesting stoichiometric relationships between insulin and C-peptide pertaining to insulin signaling. It was demonstrated that, in the presence of high insulin concentrations, C-peptide has an inhibitory effect on insulin-signaling activity. Whereas, in the presence of low insulin concentrations, C-peptide enhances insulin signaling [21, 25, 45].
It has been suggested that the enhanced insulinomimetic effects displayed by C-peptide are linked to its ability to dehexamerize natural insulin by binding of metal ions, specifically Zn2+ [22, 23, 46]. Recent data have also shown that C-peptide is internalized to the cytoplasm by endocytosis [47]. It is interesting to speculate that such a mechanism could be associated with internalization of caveolae and the co-localized insulin receptors. The earlier findings suggested a G-protein-coupled receptor mechanism, such that intracellular Ca2+ concentration evoked by C-peptide may then be explained by cross talk between insulin and G-protein-coupled receptor signaling systems [48, 49].
The effect of C-peptide on neuronal dysmetabolism
The insulin receptor (IR) originates from two mRNA forms derived from alternative splicing of exon 11. The receptor lacking exon 11 shows higher affinity for insulin, and a higher internalization rate than the isoform containing exon 11. Neuroectodermal tissue mainly contains high affinity IR [50, 51]. In peripheral nerves, the IR is localized at the nodal and paranodal axolemma, the nodal microvilli, terminal Schwann cell loops, Schmidt-Lantermann incisures, and small dorsal root ganglion (DRG) cell somata [51, 52]. Hence, at the node of Ranvier, it co-localizes with Na+-ion channels, Na+/K+-ATPase, glucose transporters, and specialized adhesive molecules like ankyrinG and caspr [53-56]. In endoneurial vessels, the IR is localized on ad- and ab-luminal endothelial cells and in endocytotic vesicles surrounding interendothelial tight junctions [51].
Initially, the expression of the IR is unaltered in the sciatic nerve of BB/Wor-rats, but increases with duration of diabetes [57]. In contrast, the expression of IR in DRGs decreases progressively with duration of diabetes [58]. These abnormalities in IR expression are normalized by C-peptide [55, 56]. By comparison, IR expression is downregulated in the sciatic nerve, and remains unaltered in DRGs in the hyperinsulinemic type 2 BBZDR/Wor-rat [14, 59].
In recent years, it became increasingly clear that impaired insulin action plays a pivotal role in the pathogenesis of DPN in type 1 diabetes [14, 15, 21, 40, 57, 58, 60]. Hence, the replacement of insulin-promoting C-peptide in type 1 diabetes provides a unique opportunity to combat neuropathic and other complications, as outlined in this Special Issue of The Review of Diabetic Studies [24, 61-64].
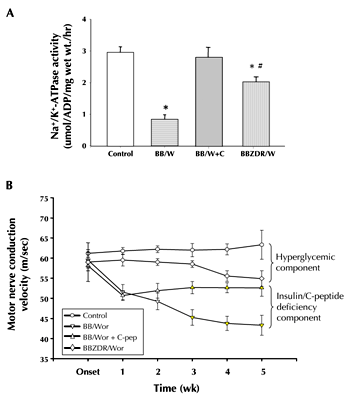
Two key abnormalities in DPN underlying the early functional nerve conduction defects are the aberrations in Na+/K+-ATPase activity and NO [65]. Impaired Na+/K+-ATPase activity has severe consequences for Na+ permeation at the node of Ranvier, and results in decreased transmembrane potential and intra-axonal Na+ accumulation causing nodal axonal swellings [66, 67]. The more severe Na+/K+-ATPase defect in type 1 diabetes (Figure 1A) can be the consequence of an insulin deficiency-mediated defect of protein kinase C activity [38, 68], and subsequent impairment of Na+/K+-ATPase phosphorylation [65, 68]. In subacutely diabetic BB/Wor-rats, C-peptide dose-dependently increases Na+/K+-ATPase activity in the sciatic nerve, diminishes nodal swelling, and increases nerve conduction velocity [38] (Figure 1B). Another mechanism, implicated in the acute nerve conduction slowing, is decreased endoneurial blood flow, secondary to impaired endothelial NO release due to impaired expression of eNOS [39, 40, 69, 70]. This has been linked both to polyol-pathway activation through a NADPH-mediated mechanism, and to changes in protein kinase activity and calcium levels [69]. Not surprisingly, in the presence of unaltered hyperglycemia, complete C-peptide replacement in type 1 diabetic BB/Wor-rats results in normalization of endoneurial blood flow and vascular conductance [39]. Interestingly, in this study, lipid peroxidation was unaffected by C-peptide, as was SOD activity, suggesting a dissociation between neurovascular deficits and oxidative stress.
 |
|
Figure 1. A: Na+/K+-ATPase activities in sciatic nerve of acutely diabetic type 1 BB/Wor-rats, C-peptide replaced BB/Wor-rats and age-matched non-diabetic control rats. For comparison, Na+/K+-ATPase activity in age- and duration-matched type 2 diabetic BBZDR/Wor-rats is included. Note markedly reduced Na+/K+-ATPase activities in BB/Wor-rats, which is fully restored in C-petide treated rats. Type 2 BBZDER/Wor rats show a modest decrease in Na+/K+-ATPase activity. * p < 0.001, # p < 0.01 vs. control rats. B: Sciatic motor nerve conduction velocity (NCV) deficits developing in acutely diabetic BB/Wor-rats, C-peptide-treated BB/Wor-rats (from 1 week duration of diabetes) and type 2 BBZDR/Wor-rats compared to age-matched non-diabetic control rats. Note the sharp and rapid decline in NCVs in type 1 BB/Wor-rats (B) corresponds to severely diminished activity of Na+/K+-ATPase (A). C-peptide-treated rats show an immediate partial but significant correction of NCV corresponding to a normalization of Na+/K+-ATPase activity. Both NCV and Na+/K+-ATPase activity defects are substantially milder in type 2 BBZDR/Wor-rats compared with type 1 BB/Wor-rats. These differences have led us to suggest a hyperglycemic component, and an insulin/C-peptide deficiency component, of the nerve conduction slowing. |
|
These early effects of C-peptide on compromised neural Na+/K+-ATPase and endoneurial vascular NO activities, underlie the corrective effects on the metabolic components of the early nerve conduction defect [13, 21, 38] (Figure 1).
The neurotrophic effects of C-peptide and apoptosis of DRGs
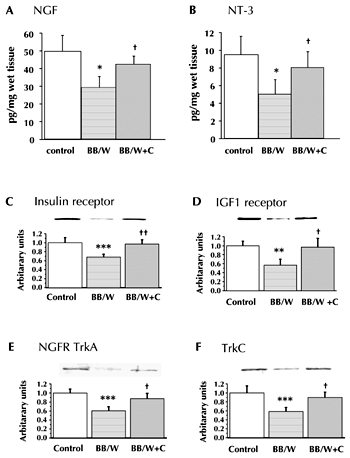
C-peptide induces activation and DNA binding of several transcription factors and exhibits a stimulatory effect on cell proliferation of neuroblastoma cells, via activation of PI-3-kinase and p38 MAP-kinase [37, 42]. Enhanced expression and translocation of NF-κB, activation of cyclic AMP response element-binding protein (CREB), and inhibition of glycogen synthase kinase 3 beta (GSK-3β) [37, 71, 72] impacts favorably on apoptotic stressors [42, 60]. Type 1 diabetes is characterized by downregulation of several neurotrophic factors. Among them are nerve growth factor (NGF), neurotrophin-3 (NT-3), and insulin-like growth factor-1 (IGF-1) and their respective receptors (Figure 2). The downregulation causes adverse effects on the synthesis and postranslational modification of axonal cytoskeletal elements, and the regenerative capacity of the diabetic nerve [57, 73, 74] (Figure 3). Besides the induction of a variety of transcription factors, C-peptide elicits the activation of early gene responses such as c-fos, c-jun, and the first waves of IGF, NGF, and NGF-p75 [14, 41, 57, 75]. This acts in a normalizing fashion on the compromised expression of neurotrophic factors and their respective receptors, and it normalizes the insulin receptor itself [57, 74] (Figure 2). There is no evidence that C-peptide per se has neurotrophic effects; the evidence points to a secondary effect mediated by its insulin-related effect.
 |
|
Figure 2. ELISA assessment of NGF (A) and NT-3 (B) in sciatic nerves of control, diabetic BB/Wor-rats and those with full substitution of C-peptide. The amounts of NGF and NT-3 are substantially decreased in diabetic rats and corrected by C-peptide to concentrations not significantly different from control rats. * p < 0.005 vs. control rats, † p < 0.05 vs. untreated BB/Wor-rats B: Protein expression of neurotrophic receptors in four mo diabetic DRG neurons. Note marked suppression of both the insulin (C), IGF-1 (D), NGF-TrkA (E) and the TrC (F) receptors in diabetic rats, and significant restorations of the expression of these receptors in diabetic rats treated with full C-peptide replacement from onset of diabetes. *** p < 0.001; ** p < 0.005 vs. control rats; †† p < 0.005; † p < 0.05 vs. untreated BB/Wor-rats. Data are compiled from references [58] and [74]. |
|
 |
|
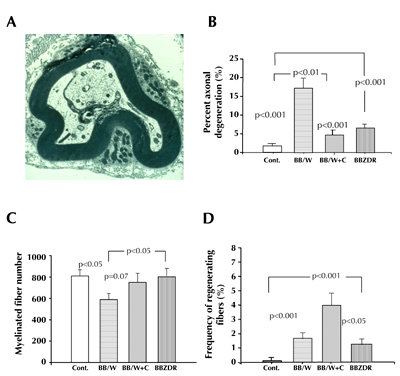
Figure 3. Impaired neurotrophic support leads to suppressed expression of tubulins and NFs and their aberrant phosphorylation [42, 73, 75]. These abnormalities lead eventually to axonal degeneration of myelinated fibers. Here depicted ultrastructurally, showing axonal atrophy and sequestration by the inner Schwann cell lip (A), which can be morphometrically assessed (B), and decreased numbers of myelinated fibers (C). ml in (A) indicate myelin lamellae. Full C-peptide substitution for eight months results in significant prevention of axonal degeneration (B), prevention of myelinated fiber loss (C) and enhances nerve fiber regeneration (D). For comparison, type 2 BBZDR/Wor-rats with the same duration of diabetes, and with the same levels of hyperglycemia, show significantly milder degrees of axonal degeneration, fiber loss (C), but the same degree of regenerative capacity (D) as non-treated BB/Wor-rats. Data are rearranged from references [28] and [85]. |
|
There are several reports on apoptotic cell death of DRG neurons as a component of diabetic neuropathy in STZ-rats [76, 77]. However, the validity of significant apoptotic cell loss of DRG neurons can be disputed, since the high apoptotic rate reported in the STZ-rat does not correspond to a comparable loss of peripheral nerve axons [78, 79]. In the BB/Wor-rat, DRGs show increased expression of proapoptotic Bax and active caspase-3. On the other hand, anti-apoptotic Bcl-xl is also increased together with unaltered NGF p75R, Fas, poly ADP-ribose polymerase-1 (PARP), and cleaved PARP [44]. This suggests that apoptotic stress occurs, but it appears to be counter-balanced by increased expression of survival factors like heat shock proteins such as HSP27 and HSP70, without significant decrease in the number of DRG neurons [44]. Instead, degenerative changes in DRG neurons, such as vacuolar subplasmolemmal changes, appear to relate to impaired Na+/K+-ATPase activity and structural changes of the Golgi complex [80].
C-peptide and axonal atrophy
Axonal atrophy with secondary axonal dying back leading to axonal loss is the very hallmark of DPN, and is more severely present in type 1 DPN [2, 28, 59] (Figure 3). Cytoskeletal neurofilaments (NFs) and tubulins are the major building blocks of the axon. Their expression and phosphorylation status impact on axonal function and size. Experimental animal models demonstrate reduced expression of NFs and tubulins in DRGs, aberrant phosphorylation, and decreased axonal transport of NFs in peripheral nerves. The three NFs (NFL, NFM, and NFH) are unique to the nervous system and assemble in a staggered fashion. They interact with microtubules, thereby providing axonal transport. Several kinases are involved in NF phosphorylation, like the cyclin-dependent kinase Cdk5, the MAP kinases Erk ½, SAPK [81], and GSKβ [82, 83].
As mentioned above, apart from the independently exerted neurotrophic effects by insulin and C-peptide, C-peptide prevents and corrects the compromised expression of IGF-1, NGF, and NT-3 and their respective receptors [74, 75] (Figure 2). These effects are mediated by the early gene regulatory functions of c-fos, c-jun, as well as by the transcript factor NF-κB [41, 42]. Downstream the correction of neurotrophic factors and their receptors result in correction of neurofilament and tubulin mRNA, and protein expression, as well as normalization of the aberrant phosphorylation of NFs [41, 84]. This has beneficial effects on both myelinated and unmyelinated axonal size and number [41, 60] (Figures 3 and 4).
 |
|
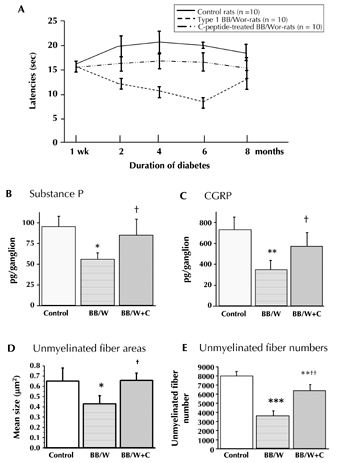
Figure 4. Diabetic BB/Wor-rats show progressive increases of hyperalgesia, as assessed from withdrawal latencies following thermal stimulation (A). Hyperalgesia is partly, but significantly, prevented in iso-hyperglycemic C-peptide replaced diabetic rats (A). These beneficial effects were underpinned by prevention of the decline in substance P (B) and CGRP (C) in small nociceptive DRG neurons, and by prevention of unmyelinated c-fiber atrophy (D), and partial but significant prevention of c-fiber loss in sural nerve (E). B and C: ** p < 0.001; * p < 0.005 vs. control rats; † p < 0.05 vs. untreated BB/Wor-rats. D and E: * p < 0.05, ** p < 0.005 and *** p < 0.001 vs. control rats; + p < 0.05 and ++ p < 0.001 vs. untreated BB/Wor-rats. Data compiled from references [58] and [74]. |
|
It is of interest to note that the upregulation of phosphorylating stress kinases, like Cdk-5, p-GSK-3β, and p42/44, exhibit specific affinities to the various NFs. Progressive upregulation of p-GSK-3β correlates significantly with progressive phosphorylation of NFH. Also, the decline in Cdk-5 expression with duration of diabetes correlates with decreased phosphorylation of NFM [84]. This emphasizes the dynamic and sequential occurrences of pathobiological mechanisms in DPN.
Morphometric assessment of C-peptide replacement
The corrective effects of C-peptide on cytoskeletal proteins are associated with prevention and correction of sural nerve myelinated fiber degeneration, fiber numbers and axonal areas, and increased frequencies of regenerating fibers in rats receiving preventional or interventional C-peptide treatment [38, 41, 58, 60, 74] (Figure 3). These beneficial effects on nerve fiber morphology translate into significant prevention and improvement of the chronic and structurally related nerve conduction velocities and hyperalgesia [58, 74, 85]. However, these corrective effects are not complete, but are only rehabilitated to the level of the much milder functional deficits in the type 2 diabetic BBZDR/Wor-rat [2, 4, 28, 59] (Figure 1). The residual defects in C-peptide-treated type 1 BB/Wor-rats can probably be ascribed to hyperglycemia-derived underlying mechanisms, such as an activated polyol-pathway and components of oxidative stress [39].
Similarly, C-peptide treatment prevents and reverses significantly degenerative changes of unmyelinated fibers, their axonal size and number, and the size of the parent nociceptive DRG neurons with prevention and significant improvements in hyperalgesia [58, 74]. These effects are discussed in the next section.
C-peptide and nociceptive neuropathy
Chronic pain is a common symptom in diabetic patients, which can severely compromise the quality of life of affected patients [1]. The underlying mechanisms are not fully understood. Current treatment options are limited and palliative in nature. They provide only limited benefits, and are sometimes associated with severe side effects [8, 86, 87]. Pain-related behavior can be assessed as exaggerated responses to painful stimuli, hyperalgesia, painful responses to normal stimuli, or allodynia.
Nociception is mediated by peripheral nociceptive fibers. Damage to unmyelinated and small myelinated Aβ fibers of small DRG neurons induce increased excitability via upregulation of tetrodotoxin-resistant Na+ channels and β-adrenergic receptors [88-90]. The high frequency of the firing pattern enhances nociception by sensitizing spinal nociceptive interneurons [91], with further modulation at the brainstem, para-aqueductal gray, and thalamic levels [92].
In the BB/Wor-rat, hyperalgesia and enhanced firing frequency of peripheral nerve occur early and progress with duration of diabetes [58, 59, 74, 88] (Figure 4A). These phenomena are substantially prevented and reversed by continuous subcutaneous administration of C-peptide [58, 74] (Figure 4A). The beneficial effects correlate with prevention and restoration of the expression of the insulin and NGF-Trk receptors (Figure 2), which are specifically enriched on small nociceptive DRG neurons and spinal nociceptive interneurons [52, 53]. Further downwards directed beneficial effects of C-peptide treatment include the restoration of nociceptive substance P, and calcitonin gene-related peptide (CGRP) synthesis [74] (Figure 4B-C). Furthermore, the progressive and duration-related atrophy of small nociceptive substance P and CGRP neurons, and the atrophy and loss of peripheral unmyelinated fiber, are significantly prevented and partially restored by C-peptide treatment [58, 74] (Figure 4D-E).
These data suggest that C-peptide prevents and modulates nociceptive symptoms, and some of the underlying mechanisms in type 1 diabetes to a significant degree. However, it should be noted, that C-peptide does not alter hyperglycemia (Table 1). Additional mechanisms related to hyperglycemia, such as spinal cyclooxygenase-2 protein and release of spinal prostaglandin E2, are likely to contribute to nociceptive neuropathy [8, 87, 92].
Regeneration and the effect of C-peptide
As mentioned above, early gene responses and subsequent perturbations of trophic factors and cytoskeletal proteins are severely affected in type 1 BB/Wor-rats [73, 75]. They are substantially milder in the iso-hyperglycemic and hyperinsulinemic type 2 diabetic counterpart, the BBDZR/Wor-rat [14, 93]. This observation suggests that insulin-deficiency is an important underlying factor in suppressed nerve fiber regeneration in type 1 DPN. C-peptide substitution to the BB/Wor-rat prior to sciatic nerve injury results in an immediate correction of early gene responses, as demonstrated by the normalization of mRNA and expression of IGI-1 protein and its receptor. It also induces sequentially c-fos and lesion-induced increases in NGF [41, 73, 75]. The attenuated upregulation of NGF-TrkA-receptor is restored by C-peptide treatment [41] (Figure 2E).
The early upregulation of NGF induces macrophage recruitment necessary for phagocytosis, secretion of interleukins, and other trophic factors [94-96]. An orderly nerve regeneration requires the amplification of neurotrophic factors in the cell somata and the availability of insulin to induce neuroskeletal protein synthesis [97-100]. C-peptide-treated rats show a timely upregulation of DRG neurotrophic factors, although the expression levels are slightly lower than in control rats [41]. Insulin and NGF upregulate NFM and NFL, and indirectly β-tubulin, by stabilizing their mRNAs [99, 101]. A proper elongation of the regenerating axon requires the upregulation of β-tubulin to precede the upregulation of NFs. In the diabetic BB/Wor-rat, the necessary downregulation of NFs does not occur and it lacks the initial upregulation of β-tubulins [41]. The normal timing and sequence in the synthesis and delivery of neuroskeletal proteins to the regenerating axons are fully restored in C-peptide-replaced type 1 diabetic rats. Normalization of these intricate relationships in C-peptide-treated rats results in a significant improvement in the elongation of regenerated fibers and normalization of regenerating fiber sizes [41].
Based on these data, it is obvious that full substitution of C-peptide has a profound normalizing effect on complicated molecular events and leads to the correct timing of neuroskeletal protein synthesis. Hence, it enables a much improved regenerative capacity in type 1 DPN.
C-peptide and nodal and paranodal degeneration
A characteristic abnormality occurring in type 1 human and experimental diabetes is the progressive nodal and paranodal degeneration, which is not seen in human or experimental type 2 diabetes [2, 79, 102, 103]. Progressive degeneration of paranodal tight junctions attaching the terminal myelin loops to the paranodal axolemma, so-called axoglial dysjunction, compromises the paranodal ion-channel barrier, and allows for lateralization of nodal α-Na+-channels [56, 103, 104] (Figure 5). This degenerative change is closely related to the chronic irreversible nerve conduction defect [56, 79, 104, 105].
 |
|
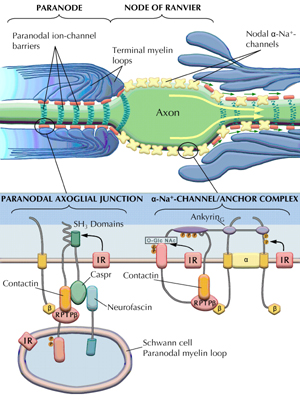
Figure 5. Schematic depiction of paranodal degeneration of the paranodal apparatus in diabetic nerve (top panel, right side) as compared to a normal paranode (top panel, left side). The molecular architecture of the paranode is shown in the lower panel, left side. Caspr interacts with RPTP-β via binding of p85 to SH3 domains. RPTP-β is a ligand of contactin. At the nodal gap (lower panel, right side) ankyrinG interacts with RPTP-β and β-Na+-channel subunits, which in turn anchor the α-Na+-channels to the nodal axolemma. |
|
At the paranode, the tight junctions connecting the myelin loops with the axolemma are made up of caspr, which interacts with other adhesive proteins such as contactin, neurofascin and receptor-like protein tyrosine phosphatase beta (RPTP-β) [106, 107] (Figures 5 and 6A). Caspr interacts with these additional proteins through binding with p85 at the SH3 domains [106, 108] (Figure 5). Interestingly, in peripheral myelinated fibers, the high affinity IR is particularly concentrated at the paranodal apparatus [51]. In type 1 DPN, caspr, contactin, and RPTP-β are significantly downregulated with a defect in p85 binding to caspr [21, 57] (Figures 5 and 7). p85, the regulatory subunit of PI3-kinase is mediated by insulin signaling. This series of events leads to disassociation of adhesive proteins and degeneration of the tight junctions, or so-called axoglial dysjunction [57]. C-peptide substitution normalizes the expression of IR, caspr, contactin, and RPTP-β, and maximizes the p85 binding to caspr in the presence of both insulin and C-peptide [57] (Figure 7). These findings are consistent with the prevention of the paranodal degenerative process by C-peptide substitution (Figure 6B), or by allogenic islet cell transplantation [79].
 |
|
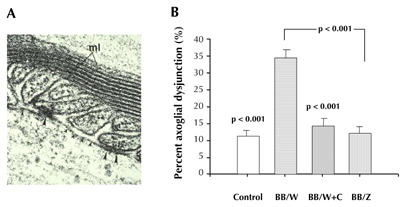
Figure 6. Disruption of the paranodal ion-channel barrier (axo-glial dysjunction) is shown ultrastructurally in A. Note the loss of electron dense tight junctions (small arrowheads), and structurally intact tight junctions (large arrowheads). Morphometric assessment of axoglial dysjunction (B) shows increased frequency in diabetic BB/Wor-rats, an increase that is fully prevented in C-peptide-replaced BB/Wor-rats. Type 2 BBZDR-rats show a normal frequency of axoglial dysjunction. Reproduced with permission from reference [57]. |
|
 |
|
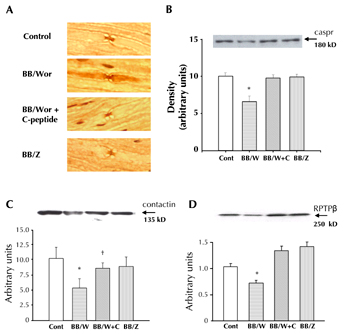
Figure 7. Immunocytochemical localization of caspr (A). Caspr is strictly localized to the paranodal areas in control, C-peptide-replaced BB/Wor-rats, and for comparison in type 2 diabetic BBZ-rats. In untreated type 1 BB/Wor-rats caspr is dispersed along the axolemma beyond the confines of the paranodal apparatus. Protein expression of caspr is decreased in diabetic BB/Wor-rats, and completely prevented in C-peptide replaced diabetic rats (B). In type 2 diabetic rats caspr is unaltered (B). The expression of contactin (C) and RPTP-β (D) was significantly decreased in BB/Wor-rats and significantly prevented by C-peptide substitution. Type 2 BBZ-rats showed no change. * p < 0.01 vs. controls; † p < 0.05 vs. untreated BB/Wor-rats. Data modified and reproduced with permission from reference [57]. |
|
At the node of Ranvier, the voltage-gated α-Na+-channels are anchored by the auxiliary β1- and β2-Na+-channel subunits, and supported by interaction with contactin and ankyrinG. The β-subunits also interact with RPTP-β, a ligand for the neuronal receptor contactin [107-109] (Figure 5). The signaling of RPTP-β is mediated via tyrosine phosphorylation sites regulated by NGF and insulin [109]. In diabetic BB/Wor-rats, the β1-Na+-channel subunit is downregulated, whereas the α-Na+-channel is unaffected, and the expression of ankyrinG is markedly suppressed. C-peptide treatment normalizes both the expression of ankyrinG and the anchoring β1-Na+-channel subunit [57]. Also, as mentioned above, both contactin and RPTP-β are normalized (Figure 7). Therefore, C-peptide stabilizes the attachment of the α-Na+-channels at the nodal axolemma and, furthermore, it prevents the breach of the paranodal ion-channel barrier. Thereby it secures the nodal localization of α-Na+-channels. These results correlate with the corrective effects of C-peptide on nodal and paranodal structural integrity [57]. Therefore, C-peptide substitution has profound beneficial effects on the chronic structurally related nerve conduction defect [13, 21, 57, 60].
C-peptide in clinical studies
The following question now arises. Do the benefits of C-peptide replacement on the multiple facets of type 1 DPN in the BB/Wor-rat, as outlined above, translate into beneficial effects on neuropathy in type 1 diabetic patients? Several controlled clinical studies have been performed to date to validate the effect of C-peptide replacement on DPN. At present, the results indicate beneficial effects on both somatic and autonomic nerve function.
In a double-blind, placebo-controlled study including 46 type 1 diabetic patients with mild DPN and reduced nerve conduction velocity (NCV), C-peptide replacement was given at 1.8 mg/day for three months. The trial showed that this treatment resulted in a 2.7 m/s increase or 80% correction of the sural NCV defect. This normalization occurred gradually and was associated with an improvement of vibration perception [110]. The results were confirmed and extended in a larger study including 139 type 1 patients with established DPN according to the Toronto-score [111]. This latter cohort included patients with advanced DPN as well as those with milder DPN and a shorter duration of diabetes. After six months of C-peptide replacement, there was a significant improvement in peak sensory NCV by 0.93 ± 0.29 m/sec. A subgroup analysis of responders with the least affected NCV at baseline showed an increase in NCV of >1 m/sec in 39% of patients treated with C-peptide. In contrast, only 5% in the placebo group showed the same change (p < 0.004). In this study, there was no statistically significant improvement in motor NCV. The reported improvements occurred in patients already on intensified insulin treatment and were independent of further improvements in glycemic control [110, 111].
Short-term infusion of C-peptide has demonstrated significant (p < 0.001) improvements in heart rate variability in type 1 diabetic patients [112]. Consistent with these data, three months replacement of C-peptide resulted in a 20% improvement in heart rate variability, whereas in placebo patients it was unchanged or slightly decreased [31, 112]. These clinical studies demonstrate a beneficial effect of C-peptide particularly in patients with mild DPN independent of glycemic control.
Discussion and conclusions
As outlined here and previously [4, 13, 15, 84], it is becoming increasingly evident that DPN is a complex, multifaceted, and highly dynamic disease process. It is also clear that DPN occurring as a consequence of type 1 and type 2 diabetes are two separate entities [4, 14, 28, 45, 59, 102, 113], although hyperglycemia is common to both disorders. The differences are even more pronounced when considering CNS complications in the two diabetes types [114-116]. It is therefore evident that biologically meaningful therapeutic approaches have to be considered separately. Consideration of the underlying DPN pathobiology can serve as a foundation for more nuanced and refined approaches to meaningful therapeutic interventions, as already demonstrated in short-term clinical trials [110, 111].
Another rarely considered aspect pertaining to the expression of type 1 DPN is the age of onset of diabetes and the potential adverse impacts of the metabolic perturbations by diabetes on normal development. In humans, the peripheral nervous system is not fully developed until well into the 20's [117, 118]. The corresponding age of full maturation of the peripheral nervous system in the rat is 6 mo [119]. Although unexplored, this may well have a particular impact in type 1 diabetes, since its global increase is occurring at an increasingly younger age [120-122]. In a recent review of data in the BB/Wor-rat, we noted that apart from progressive degenerative changes in peripheral nerves, myelinated and unmyelinated fiber numbers and sizes did not change in absolute values in diabetic animals between 2 and 10 mo of diabetes. In contrast, matched non-diabetic control rats showed a steady maturational increase in these parameters over the same age period [84], which did not occur in diabetic rats.
Based on the new findings presented in this review of DPN, and in the other reviews in this Special Issue of The Review of Diabetic Studies [62-64, 123], it is evident that type 1 diabetes is a two-hormone disorder, namely that of insulin deficiency and C-peptide deficiency. These are two hormones originating from the same pancreatic β-cells, which are immunologically destroyed in type 1 diabetes. As contested by large-scale clinical trials [9-11], control of hyperglycemia is not sufficient to prevent, or reverse, the progression of DPN in type 1 diabetes. It appears that, apart from the momentary glucose lowering effect of insulin, the myriad of additional effects of insulin and related signaling activities need to be sustained over time, which can be achieved by C-peptide, as demonstrated here. Such effects include the metabolic effects on early key culprits in DPN, like neuronal Na+/K+-ATPase activity and NO. They underlie the acute neuronal functional defects, as well as gene regulatory effects on neurotrophic factors and their receptors, via transcription factors and early gene responses. As described in this review, the beneficial effects lead to downstream corrections of neuroskeletal protein expressions, and their aberrant phosphorylation by stress kinases generated through insufficient insulin signaling. Additional downstream beneficial effects include normalization of nociceptive neuropeptides, cell-adhesive molecules, and their postranslational modifications, which are important for neuronal ion-channel barrier systems integrity. Further beneficial effects displayed by C-peptide include modulation of apoptotic stressors.
Sequentially and/or in concert, these effects prevent and improve axonal cytoskeletal integrity, promote nerve fiber regeneration, and prevent the characteristic type 1 abnormalities affecting the nodal and paranodal apparati. The effects are not likely to be mediated by C-peptide per se [25, 26]. Instead, the responsible mechanism appears to be mediated by an interaction between C-peptide and insulin. This interaction provides a more sustained overall insulin effect [21, 25, 26, 36] possibly through dehexamerization of naturally occurring insulin [22], or endosomal interaction with insulin signaling [47]. It was demonstrated that C-peptide plus insulin have additional small, but measurable, effects on blood glucose levels [22]. Patients given C-peptide require smaller insulin doses to sustain near normal glycemia [22]. Interestingly, a review of treatment charts of several series of C-peptide treated BB/Wor-rats revealed small, but significant, differences in insulin requirements to sustain the same hyperglycemic levels as in non-C-peptide-treated animals (Sima et al., unpublished data).
DPN represents a disorder for which there is presently no targeted therapy. As outlined in this review, C-peptide may provide a simple and biologically meaningful tool to fill this gap. In view of the substantial amount of data that have accumulated over the last decade, and the existence of proof-of-concept, it is somewhat surprising that the insulin manufacturing pharmaceutical industry and major diabetes granting agencies are still approaching the therapeutic potentials of C-peptide with skepticism. When one steps back and looks at the overriding basic concepts, which are now strengthened by increasing amounts of scientific and clinical data, as outlined in this issue, the rational for C-peptide replacement is obvious. Therefore, until we find the ultimate "cure" for type 1 diabetes, a disorder which is increasing globally at accelerating rates, and affecting children at an ever younger age; we could very likely, with relatively simple means by a two-hormone replacement approach, be able to alleviate the suffering of millions of young people with late complications like DPN.
Disclosures: The authors report no conflict of interests.
References
- Greene DA, Sima AA, Feldman EL, Stevens MJ. Diabetic neuropathy. In: Porte D Jr, Sherwin RS (eds). Ellenberg and Rifkin Diabetes Mellitus. 5th ed., Appleton and Lange, Stanford, 1997, pp. 1009-1076. [DOD]
- Sima AA, Kamiya H. Diabetic neuropathy differs in type 1 and type 2 diabetes. Ann NY Acad Sci 2006. 1084:235-249. [DOD] [CrossRef]
- Dyck PJ, Davies JL, Wilson DM, Service FJ, Melton LJ 3rd, O’Brien PC. Risk factors for severity of diabetic polyneuropathy: intensive longitudinal assessment of the Rochester Diabetic Neuropathy Study Cohort. Diabetes Care 1999. 22:1479-1486. [DOD] [CrossRef]
- Sima AA. Heterogeneity of diabetic neuropathy. Front Biosci 2008. 13:4809-4816. [DOD] [CrossRef]
- Sima AA, Bril V, Nathaniel V, McEwen TA, Brown M, Lattimer SA, Greene DA. Regeneration and repair of myelinated fibers in sural nerve biopsies from patients with diabetic neuropathy treated with an aldose reductase inhibitor. N Eng J Med 1988. 319:548-555. [DOD]
- Greene DA, Bril V, Sima AA. The relevance and efficacy of aldose reductase inhibitors in human diabetic neuropathy. In: Ward J, Goto Y, (eds). Diabetic Neuropathy. John Wiley and Sons Ltd., London, 1990, Chapt. 13, pp. 133-142. [DOD]
- Ziegler D, Reljanovic M, Meknert H, Gries FA. Alpha lipoic acid in the treatment of diabetic polyneuropathy in Germany, current evidence from clinical trials. Exp Clin Endocrin Diab 1999. 107:531-537. [DOD]
- Sima AA, Calvani M, Mehra M, Amato A. Acetyl-L-carnitine improves pain, vibratory perception and nerve morphology in patients with chronic diabetic peripheral neuropathy: an analysis of two randomized, placebo-controlled trials. Diabetes Care 2005. 28:96-101. [DOD] [CrossRef]
- The Diabetes Control Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. New Engl J Med 1993. 329:977-986. [DOD] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. The Lancet 1998. 352:S37-S53. [DOD] [CrossRef]
- Reichard P, Pihl M, Rosenquist U, Sule J. Complications in IDDM are caused by elevated blood glucose level: The Stockholm Diabetes Intervention Study (SDIS) at 10-year follow-up. Diabetologia 1996. 39:1483-1488. [DOD] [CrossRef]
- Cheatham B, Kahn R. Insulin action and the insulin signaling network. Endocrin Rev 1995. 16:117-142. [DOD]
- Sima AA. New insights into the metabolic and molecular basis for diabetic neuropathy. Cell Mol Life Sci 2003. 60:2445-2464. [DOD] [CrossRef]
- Pierson CR, Zhang W, Murakawa Y, Sima AAF. Early gene responses of trophic factors differ in nerve regeneration in type 1 and type 2 diabetic neuropathy. J Neuropath Exp Neurol 2002. 61:857-871. [DOD]
- Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes 2004. 53:1824-1830. [DOD] [CrossRef]
- Ishii D. Implications of insulin-like growth factors in the pathogenesis of diabetic neuropathy. Brain Res Rev 1995. 20:47-67. [DOD] [CrossRef]
- Park CR. Cognitive effects of insulin in the central nervous system. Neurosci Biobehav Rev 2001. 25:311-323. [DOD] [CrossRef]
- Luppi P, Cifarelli V, Tse H, Piganelli J, Trucco M. Human C-peptide antagonizes high glucose-induced endothelial dysfunction through the nuclear factor-kappaB pathway. Diabetologia 2008. 51:1534-1543. [DOD] [CrossRef]
- Sima AA, Zhang W, Kreipke CW, Rafols JA, Hoffman WH. Inflammation in diabetic encephalopathy is prevented by C-peptide. Rev Diabet Stud 2009. 6:37-42. [DOD] [CrossRef]
- Wiggins TD, Sullivan KA, Pop-Busui R, Amato A, Sima AAF, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes 2009. 58:1634-1640. [DOD] [CrossRef]
- Sima AA, Kamiya H. Insulin, C-peptide and diabetic neuropathy. Science Med 2004. 10:308-319. [DOD]
- Shafqat J, Melles E, Sigmudsson K, Johansson BL, Ekberg K, Alvelius G, Henriksson M, Johansson J, Wahren J, Jörnvall H. Proinsulin C-peptide elicits disaggregation of insulin resulting in enhanced physiological insulin effects. Cell Mol Life Sci 2006. 63:1805-1811. [DOD] [CrossRef]
- Meyer JA, Subasinghe W, Sima AAF, Keltner Z, Reid GE, Daleke D, Spence DM. Hyperglycemia-induced phosphatidylserine translocation is associated with zink-activated C-peptide resistance in type 2 diabetic erythrocytes. Mol BioSyst 2009. 5:1157-1162. [DOD] [CrossRef]
- Medawala W, McCahill P, Giebink A, Meyer J, Ku CJ, Spence DM. A molecular level understanding of zinc activation of C-peptide and its effects on cellular communications in the bloodstream. Rev Diabet Stud 2009. This issue. [DOD] [CrossRef]
- Grunberger G, Qiang X, Li ZG, Mathews ST, Sbriessa D, Shisheva A, Sima AAF. Molecular basis for the insulinomimetic effects of C-peptide. Diabetologia 2001. 44:1247-1257. [DOD] [CrossRef]
- Jensen ME, Messina EJ. C-peptide induces a concentration-dependent dilatation of skeletal muscle arterioles only in the presence of insulin. Am J Phys 1999. 276:H1223-H1228. [DOD]
- Mordes JP, Bortell R, Groen H, Guberski D, Rossini AA, Greiner DL. Autoimmune diabetes mellitus in the BB-rat. In: Sima AA, Shafrir E (eds.). Animal Models of Diabetes. Harwood Acad Publ, Amsterdam, 2001, pp. 1-42. [DOD]
- Sima AA, Zhang W, Xu G, Sugimoto K, Guberski DL, Yorek MA. A comparison of diabetic polyneuropathy in type-2 diabetic BBZDR/Wor-rat and in type 1 diabetic BB/Wor-rat. Diabetologia 2000. 43:786-793. [DOD] [CrossRef]
- Steiner DF, Cunningham L, Spigelman L, Aben B. Insulin biosynthesis: evidence for a precursor. Science 1967. 157:697-700. [DOD] [CrossRef]
- Steiner DR, Rubenstein AH. Proinsulin C-peptide - biological activity? Science 1997. 277:531-532. [DOD]
- Johansson BL, Borg K, Fernquist-Forbes E, Kernell A, Odergren T, Wahren J. Beneficial effects of C-peptide on incipient nephropathy and neuropathy in patients with type 1 diabetes mellitus. Diabet Med 2000. 17:181-189. [DOD] [CrossRef]
- Johansson BL, Linde B, Wahren J. Effect of C-peptide on blood flow, capillary diffusion capacity and glucose utilization in the exercising forearms of type 1 (insulin-dependent) diabetic patients. Diabetologia 1992. 35:1151-1158. [DOD] [CrossRef]
- Forst T, Kunt T, Pohlmann T, Goitom K, Engelbach M, Beyer J, Pfützner A. Biological activity of C-peptide on the skin microcirculation in patients with insulin-dependent diabetes mellitus. J Clin Invest 1998. 101:2036-2041. [DOD] [CrossRef]
- Rigler R, Pramanik A, Jonasson P, Kratz G, Jansson OT, Nygren PA, Stahl S, Ekberg K, Johansson BL, Uhlen S, Uhlen M, Jörnvall H, Wahren J. Specific binding of the proinsulin C-peptide to human cell membranes. Proc Natl Acad Sci USA 1999. 46:13318-13323. [DOD] [CrossRef]
- Othomo Y, Aperia A, Sahlgren B, Johansson BL, Wahren J. C-peptide stimulates rat renal tubular Na+, K+-ATPase activity in synergism with neuropeptide Y. Diabetologia 1996. 39:199-205. [DOD] [CrossRef]
- Grunberger G, Sima AA. The C-peptide signaling. Exp Diab Res 2004. 5:25-36. [DOD] [CrossRef]
- Li ZG, Qiang X, Sima AA, Grunberger G. C-peptide attenuates protein tyrosine phosphatase activity and enhances glycogen synthesis in L6 myoblasts. Biochem Biophys Res Com 2001. 26:615-619. [DOD]
- Zhang W, Yorek M, Pierson CR, Murakawa Y, Breidenbach A, Sima AA. Human C-peptide dose dependently prevents early neuropathy in the BB/Wor-rat. Internatl J Exp Diab Res 2001. 2(3):187-194. [DOD] [CrossRef]
- Stevens MJ, Zhang W, Li F, Sima AA. C-peptide corrects endoneurial blood flow but not oxidative stress in type 1 BB/Wor-rats. Am J Physiol 2004. 287:E497-E505. [DOD]
- Cotter MA, Ekberg K, Wahren J, Cameron NE. Effects of proinsulin C-peptide in experimental diabetic neuropathy: vascular actions and modulation by nitric oxide synthase inhibition. Diabetes 2003. 52:1812-1817. [DOD] [CrossRef]
- Pierson CR, Zhang W, Sima AA. Proinsulin C-peptide replacement in type 1 diabetic BB/Wor-rats prevents deficits in nerve fiber regeneration. J Neuropath Exp Neurology 2003. 62:765-779. [DOD]
- Li ZG, Zhang W, Sima AA. C-peptide enhances insulin-mediated cell growth and protection against high glucose induced apoptosis in SH-SY5Y cells. Diabetes Metab Res Rev 2003. 19:375-385. [DOD] [CrossRef]
- Sima AA, Li ZG. The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetes. Diabetes 2005. 54:1497-1505. [DOD] [CrossRef]
- Kamiya H, Zhang W, Sima AA. Apoptotic stress is counterbalanced by survival elements preventing programmed cell death of dorsal root ganglions in subacute type 1 diabetic BB/Wor-rats. Diabetes 2005. 54:3288-3295. [DOD] [CrossRef]
- Sima AA. Diabetic neuropathy in type 1 and type 2 diabetes and the effects of C-peptide. Neurol Sci 2004. 220:133-136. [DOD] [CrossRef]
- Meyer JA, Froelich JM, Reid GE, Karunarathne WK, Spence DM. Metal activated C-peptide facilitates glucose clearance and release of a nitric acid stimulus via the GLUT-1 transporter. Diabetologia 2008. 51:175-182. [DOD] [CrossRef]
- Luppi P, Geng X, Cifarelli V, Brain P, Trucco M. C-peptide is internalised in human endothelial and vascular smooth muscle cells via endosomes. Diabetologia 2009. 52:2218-2228. [DOD] [CrossRef]
- Kisfalvi K, Rey O, Young SH, Sinnett-Smith J, Rozengurt E. Insulin potentiates Ca2+ signaling and phosphatidylinositol 4,5-biophosphate hydrolysis induced by Gq protein-coupled receptor agonists through an mTOR-dependent pathway. Endocrinol 2007. 148:3246-3257. [DOD] [CrossRef]
- Hall KE, Liu J, Sima AAF, Wiley JW. Impaired inhibitory G protein function contributes to increased calcium currents in rats with diabetic neuropathy. J Neurophysiol 2001. 86(2):760-770. [DOD]
- Goldstein BJ, Dudley AL. The rat insulin receptor; primary structure and conservation of tissue-specific alternative messenger RNA splicing. Mol Endocrinol 1990. 4:235-244. [DOD] [CrossRef]
- Sugimoto K, Murakawa Y, Zhang WX, Xu G, Sima AA. Insulin receptor in rat peripheral nerve: its localization and alternatively spliced isoforms. Diabetes Metab Res Rev 2000. 16(5):354-363. [DOD] [CrossRef]
- Sugimoto K, Murakawa Y, Sima AA. Expression and localization of insulin receptor in rat dorsal root ganglion and spinal cord. J Peripher Nerv Syst 2002. 7(1):44-53. [DOD] [CrossRef]
- Ariyasu RG, Nichol JA, Ellisman MH. Localization of sodium/potassium adenosine triphosphate in multiple cell types of the murine nervous system with antibodies raised against the enzyme from kidney. J Neurosci 1985. 5:2581-2596. [DOD]
- Kordeli E, Lambert S, Bennett V. AnkyrinG. A new ankyrin gene with neural specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 1995. 270(5):2352-2359. [DOD] [CrossRef]
- Magnani P, Cherian PV, Gould G, Greene DA, Sima AA, Brosius FC 3rd. Glucose transporters in rat peripheral nerve: paranodal expression of GLUT1 and GLUT3. Metabolism 1996. 45(12):1466-1473. [DOD] [CrossRef]
- Cherian PV, Kamijo M, Angelides KJ, Sima AA. Nodal Na+-channel displacement is associated with nerve conduction slowing in the chronically diabetic BB/W-rat. Prevention by an aldose reductase inhibitor. J Diabetes Complications 1996. 10(4):192-200. [DOD] [CrossRef]
- Sima AA, Zhang W, Li ZG, Murakawa Y, Pierson CR. Molecular alterations underlie nodal and paranodal degeneration in type 1 diabetic neuropathy and are prevented by C-peptide. Diabetes 2004. 53:1556-1563. [DOD] [CrossRef]
- Kamiya H, Zhang W, Ekberg K, Wahren J, Sima AA. C-peptide reverses nociceptive neuropathy in type 1 diabetic BB/Wor-rat. Diabetes 2006. 55:3581-3587. [DOD] [CrossRef]
- Kamiya H, Murakawa Y, Zhang W, Sima AA. Unmyelinated fiber sensory neuropathy differs in type 1 and type 2 diabetes. Diabetes Metab Res Rev 2005. 21:448-458. [DOD] [CrossRef]
- Sima AA, Kamiya H. Is C-peptide replacement the missing link for successful treatment of neurological complications in type 1 diabetes? Curr Drug Targets 2008. 9(1):37-46. [DOD]
- Haidet J, Cifarelli V, Trucco M, Luppi P. Anti-inflammatory properties of C-peptide. Rev Diabet Stud 2009. This issue. [DOD] [CrossRef]
- Nordquist L, Wahren J. C-peptide: the missing link in diabetic nephropathy? Rev Diabet Stud 2009. This issue. [DOD] [CrossRef]
- Forst T, Hach T, Kunt T, Weber MM, Pfützner A. Molecular Effects of C-Peptide in Microvascular Blood Flow Regulation. Rev Diabet Stud 2009. This issue. [DOD] [CrossRef]
- Sima AA, Zhang W, Muzik O, Kreipke CW, Rafols JA, Hoffman WH. Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-peptide. Rev Diabet Stud 2009. This issue. [DOD] [CrossRef]
- Vague P, Coste TC, Jannot MF, Raccah D, Tsimaratos M. C-peptide, Na+, K+-ATPase and diabetes. Exp Diabesity Res 2004. 5(1):37-50. [DOD] [CrossRef]
- Brismar T, Sima AA. Changes in nodal function in nerve fibres of the spontaneously diabetic BB-Wistar rat. Potential clamp analysis. Acta Physiol Scand 1981. 113:499-506. [DOD] [CrossRef]
- Sima AA, Brismar T. Reversible diabetic nerve dysfunction. Structural correlates to electrophysiological abnormalities. Ann Neurol 1985. 18:21-29. [DOD] [CrossRef]
- Greene DA, Lattimer SA, Sima AA. Sorbitol, phosphoinositides and sodium-potassium ATPase in the pathogenesis of diabetic complications. N Engl J Med 1987. 316:599-606. [DOD]
- Stevens MJ, Dananberg J, Feldman EL, Lattimer SA, Kamijo M, Thomas TP, Shindo H, Sima AA, Greene DA. The linked roles of nitric oxide, aldose reductase and, (Na+/K+)-ATPase in the slowing of nerve conduction in the streptozotocin diabetic rat. J Clin Invest 1994. 94:853-859. [DOD] [CrossRef]
- Kutanyra T, Kimura K, Makondo K, Furuya T, Suzuki M, Yoshida T, Saito M. Proinsulin C-peptide increases nitric oxide production by enhancing mitogen-activated protein-kinase-dependent transcription of endothelial nitric oxide synthase in aortic enothelial cells of Wistar-rats. Diabetologia 2003. 46:1698-1705. [DOD] [CrossRef]
- Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey II WH, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type 1 diabetic encephalopathy. Brain 2008. 131:3311-3334. [DOD] [CrossRef]
- Hills CE, Brunskill NJ. Intracellular signaling by C-peptide. Exp Diabetes Res 2008. 2008:635158. [DOD]
- Xu G, Murakawa Y, Pierson CR, Sima AA. Altered beta-tubulin and neurofilament expression and impaired axonal growth in diabetic nerve regeneration. J Neuropath Exp Neurol 2002. 61:164-175. [DOD]
- Kamiya H, Zhang W, Sima AA. C-peptide prevents nociceptive sensory neuropathy in type 1 diabetes. Ann Neurol 2004. 56:827-835. [DOD] [CrossRef]
- Xu G, Sima AA. Altered immediate early gene expression is impaired in diabetic nerve: implications in regeneration. J Neuropath Exp Neurol 2001. 60(10):972-983. [DOD]
- Schmeichel AM, Schmetzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes 2003. 52:165-171. [DOD] [CrossRef]
- Russel JW, Sullivan KA, Windebank AJ, Herrman DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis 1999. 6:347-363. [DOD] [CrossRef]
- Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes 2003. 52:2363-2371. [DOD] [CrossRef]
- Sima AA, Zhang WX, Tai J, Tze WJ, Nathaniel V. Diabetic neuropathy in STZ-induced diabetic rat and effect of allogenic islet cell transplantation. Morphometric analysis. Diabetes 1988. 37:1129-1136. [DOD] [CrossRef]
- Kamiya H, Zhang W, Sima AA. Degeneration of Golgi and neuronal loss in dorsal root ganglia in diabetic BB/Wor-rats. Diabetologia 2006. 49(11):2763-2774. [DOD] [CrossRef]
- Terada M, Yasuda H, Kikkawa R. Delayed Wallerian degeneration and increased neurofilament phosphorylation in sciatic nerves of rats with streptozotocin-induced diabetes. J Neurol Sci 1998. 155:23-30. [DOD] [CrossRef]
- Purves T, Middlemas A, Agthong S, Jude FB, Boulton AJ, Fernyhough P, Tomlinson DR. A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB J 2001. 15:2508-2514. [DOD] [CrossRef]
- Brownlees J, Yates A, Bajaj NP, Davis D, Anderton BH, Leigh PN, Show CR, Miller CC. Phosphorylation of neurofilament heavy chain side-arms by stress activated protein kinase-1b/Jun N-terminal kinase-3. J Cell Sci 2000. 113(Pt 3):401-407. [DOD]
- Kamiya H, Zhang W, Sima AA. Dynamic changes of neuroskeletal proteins underlie impaired axonal maturation and progressive degeneration in type 1 diabetes. Exp Diabetes Res 2009. 2009:793281. [DOD]
- Sima AA, Zhang WX, Sugimoto K, Henry D, Li ZG, Wahren J, Grunberger G. C-peptide prevents and improves chronic type 1 diabetic neuropathy in the BB/Wor-rat. Diabetologia 2001. 44:889-897. [DOD] [CrossRef]
- Gilron I, Flatters SJ. Gabapentin and pregablin for the treatment of neuropathic pain: a review of laboratory and clinical evidence. Pain Res Manag 2006. 11(Suppl A):16-29. [DOD]
- Ramos KM, Jiang Y, Svensson CI, Calcutt NA. Pathogenesis of spinally mediated hyperalgesia in diabetes. Diabetes 2007. 56:1569-1576. [DOD] [CrossRef]
- Burchiel KJ, Russel LC, Lee RP, Sima AA. Spontaneous activity of primary afferent neurons in diabetic BB-Wistar rats. A possible mechanism of chronic diabetic pain. Diabetes 1985. 34:1210-1213. [DOD] [CrossRef]
- Hirade M, Yasuda H, Omatsu-Kanbe M, Kikkawa R, Kitasato H. Tetrodotoxin-resistant sodium channels of dorsal root ganglion neurons are readily activated in diabetic rats. Neuroscience 1999. 90:933-937. [DOD] [CrossRef]
- Kapur D. Neuropathic pain and diabetes. Diabetes Metab Res Rev 2003. 19:S9-S15. [DOD] [CrossRef]
- Arendt-Nielsen L, Sonnenborg FA, Andersen OK. Facilitation of the withdrawal reflex by repeated transcutaneous electrical stimulation: an experimental study on central integration in humans. Eur J Appl Physiol 2000. 81:165-173. [DOD] [CrossRef]
- Bachouja MM. Pathophysiology of neuropathic pain. In: Veves A, Malik RA (eds). Diabetic neuropathy. Clinical management. Humana Press, Totowa, NJ, 2007, pp 339-350. [DOD]
- Pierson CR, Zhang W, Murakawa Y, Sima AA. Insulin deficiency rather than hyperglycemia account for impaired neurotrophic responses and nerve fiber regeneration in type 1 diabetic neuropathy. J Neuropath Exp Neurology 2003. 62:260-271. [DOD]
- Kamijo M, Merry AC, Akdas G, Cherian PV, Sima AA. Nerve fiber regeneration following axotomy in the diabetic BB/W-rat. The effect of ARI-treatment. J Diabetes Complications 1996. 10:183-191. [DOD] [CrossRef]
- Sima AA, Merry AC, Lightle R. Impaired macrophage recruitment in axotomized diabetic nerve. Exp Clin Endocrin Diab 1997. 105:62-63. [DOD] [CrossRef]
- Sima AA, Levitan I, Ristic H, Kamijo M. Nerve fiber regeneration in the spontaneously diabetic BB/W-rat. In: Hotta N, Greene DA, Ward JD, Sima AA, Boulton AJ (eds). Diabetic Neuropathy: New Concepts and Insights. Elsevier Science B.V., Amsterdam, 1995, pp 27-36. [DOD]
- Ide C. Peripheral nerve regeneration. Neurosci Res 1996. 25:101-121. [DOD]
- Ishii DN, Glazner GW, Pu S. Role of insulin-like growth factor in peripheral nerve regeneration. Mol Neurobiol 1997. 14:67-116. [DOD] [CrossRef]
- Fernyhough P, Mill JF, Roberts JL, Ishii DN. Stabilization of tubulin mRNA's by insulin and insulin-like growth factor-1 during neurite formation. Mol Brain Res 1989. 6:109-120. [DOD] [CrossRef]
- Verge VN, Tetzlatt W, Bisby MA, Richardson PM. Influence of nerve growth factor on neurofilament gene expression in mature primary sensory neurons. J Neurosci 1990. 10:2018-2025. [DOD]
- Fernyhough P, Willars GB, Lindsay RM, Tomlinson DR. Insulin and insulin-like growth factor I enhance regeneration in cultured adult rat sensory neurons. Brain 1993. 607:117-124. [DOD] [CrossRef]
- Sima AA, Nathaniel V, Bril V, McEwen TA, Greene DA. Histopathological heterogeneity of neuropathy in insulin-dependent and non-insulin-dependent diabetes, and demonstration of axo-glial dysjunction in human diabetic neuropathy. J Clin Invest 1988. 81:349-364. [DOD] [CrossRef]
- Sima AA, Lattimer SA, Yagihashi S, Greene DA. "Axo-glial dysjunction": a novel structural lesion that accounts for poorly reversible slowing of nerve conduction in the spontaneously diabetic BB-rat. J Clin Invest 1986. 77:474-484. [DOD] [CrossRef]
- Brismar T, Sima AA, Greene DA. Reversible and irreversible nodal dysfunction in diabetic neuropathy. Ann Neurol 1987. 21:504-507. [DOD] [CrossRef]
- Sima AA, Cherian PV. Neuropathology of diabetic neuropathy and its correlations with neurophysiology. J Clin Neurosci 1997. 4:359-364. [DOD]
- Peles E, Nativ M, Lustig M, Grumet M, Schilling J, Martinez R, Plowman GD, Schlessinger J. Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J 1997. 16:978-988. [DOD] [CrossRef]
- Ratcliffe CF, Qu V, McCormick KA, Tibbs VC, Dixon JE, Schener T, Catterall WA. A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase beta. Nat Neurosci 2000. 3:437-444. [DOD] [CrossRef]
- Peles E, Nativ M, Campbell PL, Sakurai T, Martinez R, Lev S, Clary DO, Skilling J, Barnea G, Plowman GD. The carbonic ankydrase domain of receptor tyrosine phosphatase beta is a functional ligand for the axonal recognition molecule contactin. Cell 1995. 82:251-260. [DOD] [CrossRef]
- Johnson KG, Van Vactor D. Receptor protein tyrosine phosphases in nervous system development. Physiol Rev 2003. 83:1-24. [DOD]
- Ekberg K, Brismar T, Johansson BL, Jonsson B, Lindström P, Wahren J. Amelioration of sensory nerve dysfunction by C-peptide in patients with type 1 diabetes. Diabetes 2003. 52:536-541. [DOD] [CrossRef]
- Ekberg K, Brismar T, Johansson BL, Lindström P, Juntti-Berggren L, Norrby A, Berne C, Arnquist HJ, Bolinder J, Wahren J. C-peptide replacement therapy and sensory nerve function in type 1 diabetic neuropathy. Diabetes Care 2007. 30:71-76. [DOD] [CrossRef]
- Johansson BL, Borg K, Fernquist-Forbes E, Odergren T, Remahl S, Wahren J. C-peptide improves autonomic nerve function in IDDM patients. Diabetologia 1996. 39:687-695. [DOD] [CrossRef]
- Schmidt RE, Dorsey DA, Beaudet LN, Parvin CA, Zhang W, Sima AA. Experimental rat models of type 1 and type 2 diabetes differ in sympathetic neuronal dystrophy. J Neuropath Exp Neurol 2004. 63:450-460. [DOD]
- Sima AA, Kamiya H, Li ZG. Insulin, C-peptide hyperglycemia and central nervous system complications in diabetes. Europ J Pharmacology 2004. 490:187-197. [DOD] [CrossRef]
- Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 2007. 56:1817-1824. [DOD] [CrossRef]
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a life-span perspective. Lancet Neurol 2008. 7:184-190. [DOD] [CrossRef]
- Rexed B. Contributions to the knowledge of the postnatal development of the peripheral nervous system in man. Acta Psych Neurol 1944. 19(Suppl 33):71-206. [DOD]
- Jacobson M. Developmental Neurobiology. 2nd ed., Plenum Publ., New York, 1978. [DOD]
- Sima AA. Studies on fiber size in developing sciatic nerve and spinal roots in normal, undernourished and rehabilitated rats. Acta Physiol Scand 1974. 91(Suppl 1):406. [DOD]
- Harjutsalo V, Sjöberg L, Tuomiletho J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet 2008. 371:1777-1782. [DOD] [CrossRef]
- Larsson HE, Hansson G, Carlsson A, Ceder-Wall E, Jonsson B, Jönsson B, Larsson K, Lynch K, Neidernd J, Lernmark A, Ivarsson SA. DiPiS Study Group: Children developing type 1 diabetes before 6 years of age have increased linear growth independent of HLA genotypes. Diabetologia 2008. 51:1623-1630. [DOD] [CrossRef]
- Kumar P, Krishna P, Reddy SC, Gurappa M, Aravind SR, Munichoodappa C. Incidence of type 1 diabetes mellitus and associated complications among children and young adults: results from Karnataka Diabetes Registry 1995-2008. J Indian Med Assoc 2008. 106:708-711. [DOD]
- Hills CE, Brunskill NJ. C-peptide and its intracellular signaling. Rev Diabet Stud 2009. This issue. [DOD] [CrossRef]
This article has been cited by other articles:
|
Antioxidant and radical scavenging properties of garlic oil in streptozotocin-induced diabetic rats
Ashour MN, Megahed HA, Morsy SM, Eltoukhy SI, Youness ER, Habib DF, Wafai HA
Austral J Basic Appl Sci 2011. 5(10):280-286
|
|

|
Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-Peptide
Sima AA, Zhang W, Muzik O, Kreipke CW, Rafols JA, Hoffman WH
Rev Diabet Stud 2009. 6(3):211-222
|
|
|


)
)
)
)
)
)
)
)

