Review
| Rev Diabet Stud,
2010,
7(1):6-14 |
DOI 10.1900/RDS.2010.7.6 |
Functional Alterations of Proinflammatory Monocytes by T Regulatory Cells: Implications for the Prevention and Reversal of Type 1 Diabetes
Charles Sia1, Arno Hänninen2
1Vaccine Center, National Health Research Institutes, Zhunan Township, Miaoli County, Taiwan 350
2Department of Medical Microbiology & Immunology, Turku University, Finland
Address correspondence to: Charles Sia, e-mail: siady@nhri.org.tw
Manuscript submitted April 2, 2010; resubmitted April 22, 2010; accepted May 3, 2010.
Keywords: type 1 diabetes, monocyte, regulatory T cell, autoimmunity, dendritic cell, cytokines, NOD mouse
Abstract
The onset and development of type 1 diabetes (T1D) occurs in genetically predisposed individuals, and is attributed to autoimmune destruction of pancreatic β-cells involving a multitude of immune mechanisms. Defects in immune regulation may play a central role in T1D, involving impaired function and communication of both myeloid and lymphoid cells of the innate and adaptive immune compartments. Dendritic cells and regulatory T (Treg) cells are part of this network, which seem to be hampered in their quest to control and regulate tissue-destructive autoimmunity. Recent studies have shown that in vivo activated CD16- blood monocytes exhibiting proinflammatory features are present in diabetic subjects. These monocytes may govern T cell-mediated immune responses towards the development of tissue-destructive Th1 and Th17 subtypes, and give rise to inflammatory macrophages in tissues. Differential effects of cytokines IFN-γ and IL-4 in the development of inflammatory macrophages, and the distinct developmental pathways of proinflammatory or tissue-repair-associated monocytes suggest that controlling the activity of these monocytes could be part of an immune intervention strategy to prevent T1D. Similarly, strategies to target autoantigens to immature, steady-state dendritic cells could guide the immune response away from Th1 and Th17 immune effectors. This review examines potential approaches to this goal by manipulation of myeloid and lymphoid cell regulatory networks in T1D.
Abbreviations: aTreg - adaptive regulatory T cell; BDC2.5 cell - autoreactive T cell clone isolated from NOD mice; CDx - cluster of differentiation x protein (protein expressed on cells); CD11c - expressed on myeloid cells; adhesion molecule binding to fibrinogen/fibronectin; CD14 - acts as co-receptor for the detection of bacterial LPS; CD16 - low affinity Fc receptor found on monocytes and other cells; CD25 - expressed on activated regulatory T cells; CD45RA - tyrosinphosphatase, expressed by naïve lympho-cytes; CD62L - expressed on leukocytes; adhesion molecule that binds to CD34; CD80 - expressed on B cells, DCs and others; co-stimulator, ligand for; CD28 and CTLA-4; CD136 - plasma cell-surface glycoprotein, known as synde-can-1; DC - dendritic cell; DEC205 - dendritic cell receptor for endocytosis (CD205); EAE - experimental autoimmune encephalomyelitis; Fc receptor - crystallizable fragment (protein found on monocytes and other immune cells; binding specificity for part of an antibody); FoxP3 - forkhead box P3 (transcriptional activator); GM-CSF - granulocyte-macrophage colony-stimulating fac-tor; IFN - interferon; IPEX - immunodysregulation polyendocrinopathy en-teropathy X-linked syndrome (rare disease linked to the dysfunction of FoxP3; leads to dysfunction of regulatory T-cells and the subsequent autoimmunity); IL - interleukin; LPS - lipopolysaccharide; M-CSF - macrophage colony-stimulating factor; mDC - myeloid dendritic cell; MyD88 - myeloid differentiation primary response gene 88; NLR - NOD-like receptor; NOD - nonobese diabetic; nTreg cell - natural regulatory T cell; PBMC - peripheral blood mononuclear cell; pDC - plasmacytoid dendritic cell; PgE2 - prostaglandin E2; T1D - type 1 diabetes; TGF-β - transforming growth factor beta; Th - T helper cell; TLR - toll-like receptor; TNF-α - tumor necrosis factor alpha; Treg cell - regulatory T cell
Introduction
Type 1 diabetes (T1D) is a complex organ-specific autoimmune disease involving defects in immunoregulation. The disease has a relatively strong genetic association, and multiple environmental risk factors are involved in triggering its onset [1]. Different microbes, and chemical and dietary factors have been implicated either individually or in combination in causing autoimmunity to islet β-cells. In genetically predisposed individuals, such environmental factors are believed to disrupt tolerance mechanisms that normally protect islets from autoimmunity. This may lead to the emergence of diabetogenic T cells, attacking insulin-producing β-cells.
Within the context of this broadly accepted concept, intensive investigations have been made over the past several years to uncover the role played by different subsets of regulating immune cells. Thymus-derived, natural regulatory T (nTreg) cells, and adaptive Treg (aTreg) cells generated by antigenic stimulation of peripheral CD4+ lymphocytes are such subsets. They have emerged as crucial cellular gatekeepers in maintaining the integrity of the immunoregulatory circuits needed to prevent autoimmunity. Lack of these cells is due to mutations in the foxp3 gene, and results in severe multi-organ autoimmune disease including autoimmune diabetes both in humans (IPEX syndrome) and in mice (scurfy mice).
Importantly, results gathered from experimental systems showed that the generation of aTreg cells is cross-regulated by dendritic cells (DCs) [2, 3]. DCs are delicate sensors of microbial and inflammatory cues. It is the principal cell type that activates T cells of both regulatory and other subtypes. The challenge in understanding the role of DCs in the generation and activation of aTreg cells results from the remarkable degree of DC plasticity and functional heterogeneity depending on the environment where they develop and mature. Human DCs can be classified in two main subsets. The major subset contains the myeloid DCs (mDCs) that are derived from monocytes [4]. mDCs are typically located in tissues and travel via afferent lymph nodes to draining lymph nodes. They can feature two different activity forms, either low efficiency during steady-state or high efficiency during inflammatory conditions. The other subset, namely plasmacytoid DCs (pDCs), is generated from lymphoid progenitor cells. pDCs produce vast amounts of type 1 interferons (IFN) as an early response to viral infections. They do not express most of the surface markers found on mDCs [5], but they frequently express transcripts of rearranged immunoglobulin molecules. They are found in bone marrow, peripheral organs and lymphoid tissues, where they arrive mostly via high endothelial venules similarly to lymphocytes. In addition to DC and their subtypes, monocytes and macrophages are functionally heterogeneous and bear significant potential to modulate adaptive immune responses during inflammation and tissue repair.
Blood monocytes of T1D subjects exhibit proinflammatory features (increased production of interleukin-1beta (IL-1β) and IL-6), and are involved in the development of IL-17-producing lymphocytes [6]. Th17 immunity has been associated with autoimmune manifestations both in humans (multiple sclerosis, rheumathoid arthritis) and in corresponding animal models (EAE and experimental arthritides) [7]. In the nonobese diabetic (NOD) mouse, a murine model of T1D, Th17 cells have been found to be implicated in diabetes development, although several studies describe their conversion into Th1 cells before β-cell destruction. Apparently, both Th1 cells and Th17 cells can promote tissue destructive inflammation. Therefore, it is important to regulate both Th1 and Th17 cells in autoimmune diabetes. In the following sections, we discuss some emerging aspects associated with the regulation of Th1/Th17 immunity in T1D by DCs. Also, we highlight aspects of regulating cells of the monocyte/macrophage lineage within the scope of designing immune intervention strategies to halt or reverse T1D.
Blood monocytes in non-diabetic and diabetic subjects
Human monocytes are heterogeneous, comprising subpopulations at different stages of differentiation and proliferation. They all bear the lipopolysaccharide (LPS) receptor, CD14, and are further classified in two main subsets according to their surface expression of the Fcγ receptor-III CD16 [8]. The major subset contains the phagocytic CD14hiCD16- monocytes that produce high levels of proinflammatory cytokines and tumor necrosis factor (TNF-α) and interleukin-6 (IL-6), following stimulation with LPS. The CD14+CD16+ subset is heterogenous and comprises two subpopulations bearing differential levels of surface CD64 (or Fcγ receptor-I). The CD14+CD16+CD64+ subset shares the high phagocytic activity and cytokine (TNF-α and IL-6) secretion property as the CD14hiCD16- monocytes. The CD14+CD16+CD64- subset appears to resemble the alveolar CD14dimCD16+ cells, and primarily release IL-12 following LPS stimulation [9].
Both CD14+CD16+ and CD14+CD16- monocytes can be differentiated into different subpopulations of myeloid dendritic cells (mDCs). When placed in the same standard granulocyte-macrophage colony-stimulating factor (GM-CSF)- and IL-4-conditioned cultures, CD14+CD16+ monocytes are converted into CD11c+CD123- mDCs that express higher levels of the costimulatory molecule, CD80. LPS stimulation can trigger mDCs derived from the two subpopulations of monocytes to secrete comparable levels of IL-1β and IL-6. However, CD14+CD16+ monocyte-derived mDCs are found to produce significantly greater amounts of transforming growth factor beta (TGF-β), while higher levels of IL-12 are secreted by mDCs generated form CD14+CD16- monocytes [10].
The implication that blood monocytes exist in a proinflammatory state in T1D individuals results from earlier studies which analyzed serum samples from subjects at T1D onset. These samples contained significantly higher levels of monocyte-derived cytokines, such as IL-1β, IL-6, and TNF-α, than those found in healthy first-degree relatives [11-13]. Direct evidence for the proinflammatory state of blood monocytes in T1D individuals was provided by the observation that monocytes freshly fractionated from peripheral blood mononuclear cells (PBMCs) of T1D patients expressed genes of these cytokines in the absence of further stimulation [6]. IL-1β and IL-6 are involved in directing the differentiation of CD4+ lymphocytes into IL-17-producing (Th17) cells, which are associated with insulitis [14]. In this regard, IL-1β and IL-6 act on CD4+CD45RA+CD62LCD25- memory cells, and convert them into a Th17 phenotype [6].
mDC differentiation features of blood monocytes in T1D individuals
Several groups have utilized in vitro culture systems to evaluate the differentiation of blood monocytes into mDCs in T1D patients. Monocytes derived from PBMCs from patients of different age groups, and at risk children, are found to have a reduced potential to become differentiated into mDCs in GM-CSF and IL-4 conditioned cultures, as compared to those obtained from healthy controls. mDCs generated from diabetic subjects are functionally impaired as judged by their reduced capability to activate autologous T cells, and stimulate allogeneic lymphocytes [15-17]. Although mDCs from diabetic and non-diabetic individuals under similar culture conditions are not phenotypically different, mDCs generated from patients produce significantly higher levels of IL-6 following stimulation with TNF-α [18].
These results could be interpreted in the context that some of the in vivo activated proinflammatory monocytes isolated from a cohort of T1D patients did not convert into mDCs in the presence of GM-CSF and IL-4. Therefore, IL-6 detected in the culture supernatant is released by monocytes that have not fully differentiated into mDCs. Elevated levels of IL-6 could potentially result in autoregulation of activated monocytes leading them to differentiate into macrophages rather than DCs [19]. In T1D, the generation of reduced mDC numbers could then occur. This can be attributed to monocytes that do not differentiate normally into mDCs under the influence of elevated level of IL-6 produced by the supopulation of proinflammatory blood monocytes.
Functional modulation and regulation of proinflammatory monocytes for restoring tolerance in T1D
Abnormalities in blood monocytes can seriously affect DC generation, and eventually can cause disruption of the immunoregulatory circuit. On this basis, modulation of the monocytes' proinflammatory property could be considered as an essential step for immune intervention in T1D. In vitro studies showed that blood monocytes differentiate either into proinflammatory (type 1) or anti-inflammatory (type 2) macrophages [20] when exposed to microbial antigens, interferon-gamma (IFN-γ), or the Th2-associated cytokine IL-4 together with hematopoietic factors such as GM-CSF and M-CSF. The latter are known inducers of proliferation and differentiation of mononuclear phagocytic cells [21, 22].
GM-CSF-cultured monocytes share the IL-12-producing phenotype when stimulated with LPS, and they engage autologous and allogeneic CD4+ lymphocytes to trigger their activation. However, monocytes cultured in the presence of a combination of GM-CSF, IL-4, and TNF-α are modulated to differentiate in a different way. In the presence of these cytokines, CD16+ and CD16- monocytes are converted into two separate subpopulations of mDCs [10]. The CD16+-derived mDCs express higher surface levels of CD80 and CD11c than those derived from their CD16- monocyte counterpart. Upon stimulation with LPS, CD16- monocyte-derived mDCs secrete greater amounts of IL-12, while mDCs generated from CD16+ monocytes express higher levels of TGF-β. Human TGF-β is a cytokine that has been shown in vitro to be involved in the generation of tolerogenic mDCs that mediate the differentiation of naïve/inactivated human CD4+ lymphocytes into Treg cells [23-25].
On the basis of these findings, therapies to promote production and differentiation of CD16+ monocytes and repression of CD16- inflammatory monocytes, or therapies to steer differentiation of CD16+ monocytes into TGF-β-producing mDCs, could be options for immune intervention in T1D. These monocyte subsets derive from separate developmental pathways, and do not appear to be interchangeable. Therefore, it might be possible to influence their balance during development through modulation of growth factor and cytokine balances. In vitro expansion of TGF-β-producing mDCs might be possible through a combination of GM-CSF, IL-4, and TNF-α. The recombinant form of GM-CSF has gained previous regulatory approvals in human trials. Whether a safe dose of the cytokines may act on proinflammatory blood monocytes in T1D subjects to redirect their differentiation into non-pathogenic or IL-10-producing mDCs is unclear. In any case, these criteria would set the benchmark to evaluate the feasibility of this regimen.
M-CSF-exposed monocytes are differentiated into macrophages that express the scavenger receptor, CD136. These CD136+ macrophages bear anti-inflammatory features, as shown by their IL-10 secretion property, upon exposure to LPS. Interestingly, M-CSF-modulated monocytes possess regulatory properties as they can interact with CD4+ lymphocytes and convert them into CD4+D25+FOXP3+ Treg cells [26]. Furthermore, studies have shown that CD4+CD25+FOXP3+ Treg cells interact with blood monocytes to convert them into macrophages resembling the characteristics of M-CSF-derived macrophages. This event is attributed to IL-10, IL-4, and IL-13 produced by the CD4+CD25+FOXP3+ Treg cells following engagment with the monocytes [27].
In T1D development, autoreactive CD4+ cells bearing the Th1 phenotype are implicated in causing islet damage, either directly, or indirectly. Nitric oxide or oxygen radicals released by effector lymphocytes are shown to be toxic to islet β-cells. Furthermore, IL-2 and IFN-γ produced by islet-specific Th1 cells can potentiate the generation of diabetogenic CD8+ T cells that infiltrate the pancreatic islets, and attack insulin-secreting β-cells [28, 29]. IL-10 is known to exhibit a key role in suppressing Th1 responses [30]. The cytokine is also primarily involved in the induction of adaptive Treg cells from peripheral CD4+ lymphocytes [31].
Within the context of this information, it is not clear whether serum M-CSF level is elevated in T1D patients. The range of M-CSF measured in the serum samples of normal human subjects is between 700-900 units per ml [32]. In cancer trials, infusion of recombinant M-CSF at a dose that does not cause hepatic toxicity, led to the expansion of circulatory phagocytes [33-36]. These reports may serve as a reference for the rationale of evaluating M-CSF-based therapy in the immune intervention of T1D. In developing a possible therapy, it will be important to clarify whether administration of a permissible dose of the cytokine is capable of directly modulating the proinflammatory function of the monocytes in T1D subjects. In addition, it will be necessary to clarify if the cytokine can guide the differentiation pathway of the blood monocytes to become IL-10-producing macrophages able to promote the generation of Treg cells in restoring tolerance.
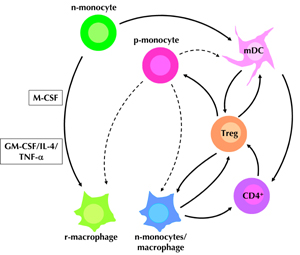
Various experimental systems showed that there is a cross-regulatory relationship between DCs and Treg cells [2, 3]. Observations by Tiemessan et al. revealed that cross-regulation also occurs between anti-inflammatory macrophages and Treg cells. In fact, macrophages in the gut lamina propria of mice were recently shown to induce Foxp3-expressing Treg cells and to suppress the induction of Th17 cells by neighboring DCs [37]. Production of immunomodulatory cytokines such as IL-10 and TGF-β by Treg cells, in turn, suppresses monocyte maturation and production of TNF-α, IFN-γ and IL-6 [38]. This suggests that Treg cells are also instrumental in controlling proinflammatory monocytes in T1D individuals. Figure 1 summarizes the discussed approaches based on modulation of the proinflammatory monocytes with cytokines to steer their differentiation into non-pathogenic or regulatory macrophages/DCs.
 |
 |
Figure 1. Conceptual approaches to redirect the differentiation of proinflammatory blood monocyes in T1D subjects. Normal blood monocytes (n-monocytes) cultured in the presence of GM-CSF and IL-4 would differentiate into CD11c+ mDCs. Subpopulations of the normal blood monocytes upon exposure to a combination of GM-CSF, IL-4, and TNF-α, or M-CSF are shown to differentiate and acquire TGF-β- and IL-10-expressing phenotypes, respectively. The regulatory monocytes/macrophages (r-monocytes/macrophages) and DCs generated could then cross-regulate the functional activity of T regulatory (Treg) cells to maintain the integrity of the immunoregulatory circuits and to prevent T1D development. However, it is unclear whether the proinflammatory monocytes found in T1D subjects could be steered to differentiate along these pathways (indicated by dashed lines). If they do, the information would be useful to devise strategies for immune intervention in T1D. |
|
Cross-talk between microbes and myeloid cells: relevance for diabetes?
Monocytes as well as other myeloid cells can recognize a variety of microbe-associated molecular patterns. They respond to microbial stimuli via their pattern recognition receptors such as toll-like receptors (TLRs) and NOD-like receptors (NLRs). Thus, microbes, or the collection of different microbial species, lining the surface of mucosal epithelium in gastrointestinal, respiratory, and genitourinary tracts are an important environmental factor and affect the inflammatory vs. tolerogenic balance in these tissue compartments.
NOD mice develop diabetes most reliably when they are kept in an environment with a low microbial load [39]. MyD88 is a key element in innate immune signaling via TLRs, and is supposedly involved in the cross-talk of innate immune cells with microbes lining mucosal surfaces. Recently, it was discovered that NOD mice deficient of MyD88 harbor gut microbiota which differs from wild type (i.e. MyD88 competent) NOD mice and confers partial protection against diabetes development [40]. The idea that the cross-talk between gut microbes and the immune system is disturbed in diabetes is also supported by the observations that NOD mice kept under conditions allowing high diabetes incidence develop an inflammatory milieu in the gut soon after weaning [41], and show aberrancies in the antigen-presenting function of innate B lymphocytes [42]. These observations are dependent on the nutrition and type of gut microbiota in the mouse colony. This suggests a role for microbe-associated factors in regulation of diabetogenesis, as was also found in the MyD88-deficient NOD mice.
In the human setting, aberrant immunologic activity in the small intestine in children with T1D, and microbial colonization of the gut in infants has been implicated in the subsequent regulation of immune responses elicited against food antigens [43, 44]. In infants suffering from atopy, administration of lactobacilli after birth was shown to reduce symptoms for an extended follow-up period. It is not clear whether regulation of immune responses to autoantigens such as insulin could be modulated by microbial adjuvants (or probiotics). However, the findings imply that administration of selected probiotic bacteria (perhaps as part of autoantigen feeding regimens) to induce Treg cells may be an effective and safe regimen that should be investigated for its applicability in the immune intervention of T1D.
Induction of Treg cells by targeted presentation of autoantigen to dendritic cells
Inflammatory stimuli derived from tissue or secreted by various leukocyte subtypes (e.g. natural killer cells) present in the lymph nodes shape the local milieu during activation of antigen-specific T cells. Inflammatory stimuli can be created by IL-12, IFN-γ, IL-1β, and IL-6. These cytokines induce maturation and activation of DCs, and favor differentiation of responding T cells into Th1, and/or Th17 subsets. In the absence of inflammatory stimuli, DCs remain in a "steady-state", display immature phenotype, and typically induce T cell tolerance instead of productive immune response (reviewed in Mueller, 2010 [45]).
Peripheral tissue antigens such as islet antigens are normally presented by DCs in the absence of inflammatory stimuli. This typically leads to either deletion of antigen-specific T cells or to induction of Treg cells (reviewed in Steinman and Nussenzweig, 2002 [46]). DCs cultured in the presence of immunosuppressive agents such as corticosteroids, 1,25-dihydroxyvitamin D, or prostaglandin E2 (PgE2) are rendered immature. Adoptive transfer of such DCs may be exploited to induce tolerance in transplantation medicine and in autoimmune diseases (reviewed in Morelli and Thomson, 2007 [47]). However, it is also possible to target antigen directly to steady-state DCs in vivo by taking advantage of their expression of the endocytosis receptor DEC205 (CD205). Coupling of antigen to a monoclonal antibody against DEC205 can target it to the surface of immature DC. Initially, the feasibility of this approach was shown by studying peripheral deletion of CD8 T cells specific for a model antigen, ovalbumin [48]. More recently, the role of DEC205+ DCs in induction of Foxp3 Treg cells was established [49]. Due to the selective role of DEC205+ DC in Treg cell induction, it is an attractive idea to target autoantigens to DEC205+ DCs to induce de novo Foxp3+ Treg cells.
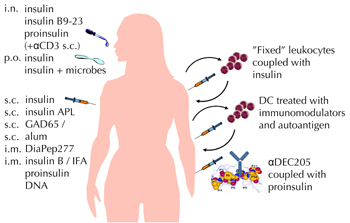
In this issue of The Review of Diabetic Studies, Petzold and coworkers report their seminal studies in the NOD mouse model to induce islet-specific Treg cells via in vivo targeting of autoantigen to immature DCs [50]. Previously, they showed that coupling antigen to fusion antibodies against the DEC205 endocytic receptor on steady-state DCs induces Treg cells in vivo [51]. This strategy had potential benefits in preventing disease progression. Now they went a step further. They took the approach of coupling autoantigen (proinsulin or a mimotope of BDC2.5 cells) similarly to anti-DEC205 antibody, and studied the feasibility of inducing islet-specific Foxp3 Treg cells during ongoing autoimmunity [50]. It showed that BDC2.5-derived Foxp3 Treg cells persisted over time in mice receiving injections of autoantigen-mimotope coupled to DEC205. Also, the anti-DEC-205-mediated targeting of whole proinsulin in prediabetic NOD mice substantially reduced the incidence of diabetes. As the number of mice studied was small, these findings on the potential efficacy in suppressing diabetes await confirmation by subsequent studies. However, while other regimens of administering autoantigen under tolerance-promoting conditions do not yield more convincing results than we currently have, targeting autoantigen to DEC205+ DC remains an interesting approach that deserves further study. A summary of potential approaches of inducing antigen-specific tolerance in T1D (including approaches mainly tested in other autoimmune diseases and transplantation medicine) is depicted in Figure 2.
 |
|
Figure 2. Approaches of antigen-specific immunotherapy for T1D. Education of the immune system to tolerate islet autoantigens by inducing passive (anergy of pathogenic leukocytes) or active (promotion of regulatory T cells) tolerance is a long sought treatment for T1D. Alum: aluminium dihydroxide. IFA: incomplete Freund’s adjuvant. APL: altered peptide ligand. DiaPep277: a peptide derived from heat shock protein 60. Dosing regimens: i.n. (intranasal), p.o. (peroral), s.c. (subcutaneous), i.m. (intramuscular). αCD3: modified antibody against T-lymphocyte CD3 receptor. “Fixed” leukocytes: ethylenecarbodiimide-treated, killed leukocytes to which autoantigen is covalently attached. DC: DC differentiated in vitro from blood monocytes in the presence of immunomodulators such as corticosteroids, prostaglandin E2 or vitamin D. αDEC205: antibody against dendritic cell endocytosis receptor DEC205. Data from both animal models and clinical trials are included (see text for references and details). |
|
Conclusions
The findings that in vivo activated blood monocytes displaying proinflammatory features are present in the circulation of T1D patients calls for measures to control their activities and to restore a balanced immunity in prediabetes. Monocytes clearly have their role in balancing effector and regulatory T cell induction.
Yet, efforts to utilize DCs to restore immune tolerance must also be continued. The work by Petzold et al. together with the increasing insight into peripheral tolerance mechanisms demonstrate the potential of DC modulation to induce Treg cells [45, 47, 50, 51]. We expect a continued research interest in testing these and conceptually similar approaches (reviewed in Luo et al., 2010 [52]) for their applicability to regulate autoimmunity in animal models of type 1 diabetes and in clinical trials involving human subjects at risk of developing type 1 diabetes.
Disclosures (conflict of interests statement): The authors report no conflict of interests.
References
- Biros E, Jordan MA, Baxter AG. Genes mediating environment interactions in type 1 diabetes. Rev Diabet Stud 2005. 2(4):192-207. [DOD] [CrossRef]
- Hubert P, Jacobs N, Caberg JH, Boniver J, Delvenne P. The cross-talk between dendritic and regulatory T cells: good or evil? J Leukoc Biol 2007. 82(4):781-794. [DOD]
- Chang HW, Chow YH, Chong P, Sia C. The cross-regulatory relationship between human dendritic and regulatory T cells and its role in type 1 diabetes mellitus. Rev Diabet Stud 2007. 4(2):68-76. [DOD] [CrossRef]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998. 392(6673):245-252. [DOD] [CrossRef]
- McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol 2005. 79(1):17-27. [DOD] [CrossRef]
- Bradshaw EM, Raddassi K, Elyaman W, Orban T, Gottlieb PA, Kent SC, Hafler DA. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol 2009. 183(7):4432-4439. [DOD] [CrossRef]
- Cooke A. Th17 cells in inflammatory conditions. Rev Diabet Stud 2006. 3(2):72-75. [DOD] [CrossRef]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol 2005. 5(12):953-964. [DOD] [CrossRef]
- Skrzeczynska-Moncznik J, Bzowska M, Loseke S, Grage-Griebenow E, Zembala M, Pryjma J. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand J Immunol 2008. 67(2):152-159. [DOD] [CrossRef]
- Sanchez-Torres C, Garcia-Romo GS, Cornejo-Cortes MA, Rivas-Carvalho A, Sanchez-Schmitz G. CD16+ and CD16- human blood monocyte subsets differentiate in vitro to dendritic cells with different abilities to stimulate CD4+ T cells. Int Immunol 2001. 13(12):1571-1581. [DOD] [CrossRef]
- Hussain MJ, Maher J, Warnock T, Vats A, Peakman M, Vergani D. Cytokine overproduction in healthy first degree relatives of patients with IDDM. Diabetologia 1998. 41(3):343-349. [DOD] [CrossRef]
- Hussain MJ, Peakman M, Gallati H, Lo SS, Hawa M, Viberti C, Watkins PJ, Leslie RD, Vergani D. Elevated serum levels of macrophage-derived cytokines precede and accompany the onset of IDDM. Diabetologia 1996. 39(1):60-69. [DOD]
- Dogan Y, Akarsu S, Ustundag B, Yilmaz E, Gurgoze MK. Serum IL-1beta, IL-2, and IL-6 in insulin-dependent diabetic children. Mediators Inflamm 2006. 2006(1):59206. [DOD]
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 2007. 8(9):942-949. [DOD] [CrossRef]
- Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet 1995. 345(8948):491-492. [DOD] [CrossRef]
- Takahashi K, Honeyman MC, Harrison LC. Impaired yield, phenotype, and function of monocyte-derived dendritic cells in humans at risk for insulin-dependent diabetes. J Immunol 1998. 161(5):2629-2635. [DOD]
- Skarsvik S, Tiittanen M, Lindstrom A, Casas R, Ludvigsson J, Vaarala O. Poor in vitro maturation and pro-inflammatory cytokine response of dendritic cells in children at genetic risk of type 1 diabetes. Scand J Immunol 2004. 60(6):647-652. [DOD] [CrossRef]
- Zacher T, Knerr I, Rascher W, Kalden JR, Wassmuth R. Characterization of monocyte-derived dendritic cells in recent-onset diabetes mellitus type 1. Clin Immunol 2002. 105(1):17-24. [DOD] [CrossRef]
- Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol 2000. 1(6):510-514. [DOD] [CrossRef]
- Verreck FA, de Boer T, Langenberg DM, van der Zanden L, Ottenhoff TH. Phenotypic and functional profiling of human proinflammatory type-1 and anti-inflammatory type-2 macrophages in response to microbial antigens and IFN-gamma- and CD40L-mediated costimulation. J Leukoc Biol 2006. 79(2):285-293. [DOD] [CrossRef]
- Fixe P, Praloran V. M-CSF: haematopoietic growth factor or inflammatory cytokine? Cytokine 1998. 10(1):32-37. [DOD]
- Motoyoshi K, Yoshida K, Hatake K, Saito M, Miura Y, Yanai N, Yamada M, Kawashima T, Wong GG, Temple PA, et al. Recombinant and native human urinary colony-stimulating factor directly augments granulocytic and granulocyte-macrophage colony-stimulating factor production of human peripheral blood monocytes. Exp Hematol 1989. 17(1):68-71. [DOD]
- Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol 2004. 172(8):4676-4680. [DOD]
- Rao PE, Petrone AL, Ponath PD. Differentiation and expansion of T cells with regulatory function from human peripheral lymphocytes by stimulation in the presence of TGF-beta. J Immunol 2005. 174(3):1446-1455. [DOD]
- Horwitz DA, Zheng SG, Wang J, Gray JD. Critical role of IL-2 and TGF-beta in generation, function and stabilization of Foxp3+CD4+ Treg. Eur J Immunol 2008. 38(4):912-915. [DOD] [CrossRef]
- Savage ND, de Boer T, Walburg KV, Joosten SA, van Meijgaarden K, Geluk A, Ottenhoff TH. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T cells, which suppress via membrane-bound TGFbeta-1. J Immunol 2008. 181(3):2220-2226. [DOD]
- Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A 2007. 104(49):19446-19451. [DOD] [CrossRef]
- Atkinson MA, Maclaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med 1994. 331(21):1428-1436. [DOD] [CrossRef]
- Durinovic-Bello I. Autoimmune diabetes: the role of T cells, MHC molecules and autoantigens. Autoimmunity 1998. 27(3):159-177. [DOD] [CrossRef]
- Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001. 19:683-765. [DOD] [CrossRef]
- Levings MK, Sangregorio R, Galbiati F, Squadrone S, de Waal Malefyt R, Roncarolo MG. IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol 2001. 166(9):5530-5539. [DOD]
- Suehiro A, Imagawa T, Hosokawa H, Suehiro M, Ohe Y, Kakishita E. Age related elevation of serum macrophage colony stimulating factor (M-CSF) level. Arch Gerontol Geriatr 1999. 29(1):13-20. [DOD] [CrossRef]
- Nemunaitis J, Meyers JD, Buckner CD, Shannon-Dorcy K, Mori M, Shulman H, Bianco JA, Higano CS, Groves E, Storb R, et al. Phase I trial of recombinant human macrophage colony-stimulating factor in patients with invasive fungal infections. Blood 1991. 78(4):907-913. [DOD]
- Sanda MG, Yang JC, Topalian SL, Groves ES, Childs A, Belfort R Jr, de Smet MD, Schwartzentruber DJ, White DE, Lotze MT, et al. Intravenous administration of recombinant human macrophage colony-stimulating factor to patients with metastatic cancer: a phase I study. J Clin Oncol 1992. 10(10):1643-1649. [DOD]
- Zamkoff KW, Hudson J, Groves ES, Childs A, Konrad M, Rudolph AR. A phase I trial of recombinant human macrophage colony-stimulating factor by rapid intravenous infusion in patients with refractory malignancy. J Immunother 1991. 11(2):103-110. [DOD] [CrossRef]
- Redman BG, Flaherty L, Chou TH, Kraut M, Martino S, Simon M, Valdivieso M, Groves E. Phase I trial of recombinant macrophage colony-stimulating factor by rapid intravenous infusion in patients with cancer. J Immunother 1992. 12(1):50-54. [DOD] [CrossRef]
- Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol 2007. 8:1086-1094. [DOD] [CrossRef]
- Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, Bijlsma JW, Lafeber FP. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol 2005. 66:222-230. [DOD] [CrossRef]
- Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 1997. 7(6):727-738. [DOD] [CrossRef]
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 2008. 455(7216):1109-1113. [DOD] [CrossRef]
- Alam C, Valkonen S, Palagani V, Jalava J, Eerola E, Hänninen A. Inflammatory tendencies and over production of IL-17 in the colon of young NOD mice are counteracted with diet change. Diabetes 2010. In press. [DOD]
- Alam C, Valkonen S, Ohls S, Tornqvist K, Hanninen A. Enhanced trafficking to the pancreatic lymph nodes and auto-antigen presentation capacity distinguishes peritoneal B lymphocytes in non-obese diabetic mice. Diabetologia 2010. 53:346-355. [DOD] [CrossRef]
- Vaarala O. The gut immune system and type 1 diabetes. Ann N Y Acad Sci 2002. 958:39-46. [DOD]
- Westerholm-Ormio M, Vaarala O, Pihkala P, Ilonen J, Savilahti E. Immunologic activity in the small intestinal mucosa of pediatric patients with type 1 diabetes. Diabetes 2003. 52(9):2287-2295. [DOD] [CrossRef]
- Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol 2010. 11:21-27. [DOD] [CrossRef]
- Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A 2002. 99:351-358. [DOD] [CrossRef]
- Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol 2007. 7:610-621. [DOD] [CrossRef]
- Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med 2002. 196:1627-1638. [DOD] [CrossRef]
- Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, Nussenzweig MC, Steinman RM. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol 2008. 181:6923-6933. [DOD]
- Petzold C, Riewaldt J, Koenig T, Schallenberg S, Kretschmer K. Dendritic cell-targeted pancreatic beta-cell antigen leads to conversion of self-reactive CD4+ T cells into regulatory T cells and promotes immunotolerance in NOD mice. Rev Diabet Stud 2010. This issue. [DOD] [CrossRef]
- Kretschmer K, Heng TS, von Boehmer H. De novo production of antigen-specific suppressor cells in vivo. Nat Protoc 2006. 1(2):653-661. [DOD] [CrossRef]
- Luo X, Herold KC, Miller SD. Immunotherapy of type 1 diabetes: where are we and where should we be going? Immunity 2010. 32:488-499. [DOD]
This article has been cited by other articles:

|
The Association between Autoimmune Disorders and Chronic Granulomatous Disease
Neelu K, Gisoo G
Pediatr Allergy Immunol Pulmonol 2014. 27(3):147-150
|
|
|
The effect of metronomic versus standard chemotherapy on the regulatory to effector T-cell equilibrium in cancer patients
Koumarianou A, Christodoulou MI, Patapis P, Papadopoulos I, Liakata E, Giagini A, Stavropoulou A, Poulakaki N, Tountas N, Xiros N, Economopoulos T, Pectasides D, Tsitsilonis OE, Pappa V
Exp Hematol Oncol 2014. 3(1):3
|
|

|
Up-regulation of fas and fasL pro-apoptotic genes expression in type 1 diabetes patients after autologous haematopoietic stem cell transplantation
de Oliveira GL, Malmegrim KC, Ferreira AF, Tognon R, Kashima S, Couri CE, Covas DT, Voltarelli JC, de Castro FA
Clin Exp Immunol 2012. 168(3):291-302
|
|
|
The biological activity of macrophages in health and disease
Nazimek K, Bryniarski K
Postepy Hig Med Dosw (Online) 2012. 66:507-520
|
|

|
The potential of incretin-based therapies in type 1 diabetes
Suen CS, Burn P
Drug Discov Today 2012. 17(1-2):89-95
|
|
|
Abundant CD4 Th-2 cytokine stimulation by medicinal plant Pongamia pinnata Linn. on human peripheral blood mononuclear cell (PBMC)
Manikannan M, Durgadevi P, Subramaniyan S, Manickan E
Int J Plant Physiol Biochem 2012. 4(2):27-32
|
|
|


)
)

