Review
| Rev Diabet Stud,
2011,
8(3):293-306 |
DOI 10.1900/RDS.2011.8.293 |
Physiology of Incretins in Health and Disease
Carolyn F. Deacon1, Bo Ahrén2
1Department of Biomedical Sciences, University of Copenhagen, DK-2200 Copenhagen N, Denmark
2Department of Clinical Sciences Lund, Lund University, SE-221 84 Lund, Sweden
Address correspondence to: Carolyn F. Deacon, e-mail: deacon@sund.ku.dk
Manuscript submitted September 1, 2011; resubmitted October 6, 2011; accepted October 10, 2011.
Keywords: DPP-4, GIP, GLP-1, glucose-dependent insulinotropic polypeptide, glucagon-like peptide-1, glycemic control, gut hormone, incretin, obesity, type 2 diabetes
Abstract
The incretin hormones, glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1), are gut peptides which are secreted by endocrine cells in the intestinal mucosa. Their plasma concentrations increase quickly following food ingestion, and carbohydrate, fat, and protein have all been shown to stimulate GLP-1 and GIP secretion. Although neural and hormonal mechanisms have also been proposed to regulate incretin hormone secretion, direct stimulation of the enteroendocrine cells by the presence of nutrients in the intestinal lumen is probably the most important factor in humans. The actions of the incretin hormones are crucial for maintaining normal islet function and glucose homeostasis. Furthermore, it is also now being recognized that incretin hormones may have other actions in addition to their glucoregulatory effects. Studies have shown that GLP-1 and GIP levels and actions may be perturbed in disease states, but interpretation of the precise relationship between disease and incretins is difficult. The balance of evidence seems to suggest that alterations in secretion and/or action of incretin hormones arise secondarily to the development of insulin resistance, glucose intolerance, and/or increases in body weight rather than being causative factors. However, these impairments may contribute to the deterioration of glycemic control in diabetic patients.
Abbreviations: AMP - adenosine monophosphate; ATP - adenosine triphosphate; BMI - body mass index; cAMP - cyclic adenosine monophosphate; CVD - cardiovascular disease; DPP-4 - dipeptidyl peptidase-4; GABA - gamma-aminobutyric acid; GIP - glucose-dependent insulinotropic polypeptide; Glc - glucose; GLP-1 - glucagon-like peptide-1; GPR - G-protein-coupled receptors; K - potassium; Na - sodium; NEFA: non-esterified fatty acid; PC1/3 - prohormone convertase 1/3; SGLT-1 - sodium-glucose cotransporter 1; TGR5 - G-protein-coupled bile acid receptor 5 (also called Gpbar-1)
Introduction
Incretin hormones are gut peptides secreted in response to nutrient ingestion, which play a key role in the regulation of islet function and blood glucose levels. In humans, the major incretin hormones are glucagon-like peptide (GLP)-1 and glucose-dependent insulinotropic polypeptide (GIP), and together they fully account for the incretin effect [1]. The incretin effect is defined as the phenomenon whereby orally ingested glucose elicits a much greater insulin response than that obtained when glucose is infused intravenously to give identical blood glucose levels (the so-called isoglycemic glucose infusion). Depending on the size of the stimulus, the incretin effect can account for up to 70% of glucose-induced insulin secretion in healthy humans [2]. It is explained by the fact that oral, but not intravenous, glucose stimulates the release of the incretin hormones which then enhance glucose-stimulated insulin secretion.
Interestingly, a similar phenomenon has recently been reported for lipids, with oral ingestion being associated with increased insulin (and incretin hormone) responses compared with matched intravenous lipid infusions [3]. GIP is produced in the K-cells, located predominantly in the more proximal parts of the small intestine, with highest density in the duodenum. In contrast, GLP-1 is produced by the more distally situated L-cells, primarily in the ileum, and is also found in high density in the colon [4]. However, both cell types can be found throughout the whole intestine, and there is also evidence that a population of cells exists in which both GLP-1 and GIP are co-localized [5].
Both K- and L-cells are "open-type" endocrine cells located in the intestinal mucosa, meaning that they can be influenced by direct contact with nutrients derived from food ingestion. It has been proposed, however, that other mechanisms are also involved in regulating the secretion of GLP-1 and GIP. Once released, both hormones are rapidly degraded by the enzyme dipeptidyl peptidase 4 (DPP-4) [6, 7]. The discovery that DPP-4 has a key role in the degradation of GLP-1 has led to the development of DPP-4 inhibitors as therapeutic agents in the management of type 2 diabetes [8, 9].
Incretin hormone biosynthesis and metabolism
GLP-1 is derived from the post-translational processing of the proglucagon gene product by prohormone convertase 1/3 (PC-1/3) in the intestinal L-cells. This process also results in production of the related peptide GLP-2. In the pancreatic α-cell, the action of PC-2 gives rise to glucagon [10]. However, recent studies suggest that, under some circumstances, small amounts of GLP-1 may also be produced in the α-cell, with isolated rat and human islets being shown to contain GLP-1 following exposure to high glucose concentrations [11, 12]. In the L-cell, proglucagon processing generates a 31-residue peptide, designated GLP-1(7-37). This peptide can be amidated at the C-terminal glycine residue to give rise to a second form, GLP-1(7-36)NH2. In humans, the amidation process is efficient, so that the majority of the peptide released from the L-cell is in the amidated form [13].
In the case of GIP, PC-1/3 processing of pro-GIP within the K-cells results in the formation of a single bioactive 42-residue peptide [14]. As for GLP-1, there is some evidence to suggest that α-cells may also be able to produce small amounts of GIP, which undergoes processing by PC-2 to form a C-terminally truncated peptide, GIP(1-30) [15]. In the intestinal enteroendocrine cells, both GLP-1 and GIP are stored as intact peptides within secretory granules until they are secreted. Once released, N-terminal degradation by DPP-4 forms the metabolites, GLP-1(9-37)/GLP-1(9-36)NH2 and GIP(3-42), which account for the majority of the circulating forms of the two incretins [6, 7]. This degradation is a rapid process, giving the intact peptides short half-lives of only 1-2 minutes (GLP-1) [16, 17] and 2-3 minutes (GIP) [7, 18].
Incretin hormone secretion
Direct luminal stimulation of enteroendocrine cells
Incretin hormone release is strongly correlated with food intake [19, 20]. Plasma concentrations rise quickly; increases are already apparent within minutes after nutrient ingestion, and remain above basal levels for several hours [21]. The driving factor for stimulating incretin hormone secretion seems to be the presence of nutrients within the intestinal lumen rather than distension of the stomach, since ingestion of a water load only results in a minimal increase in GLP-1 and GIP concentrations [22]. The release of GLP-1 is related to the rate of gastric emptying and the appearance of nutrients in the intestinal lumen [23, 24]. In contrast, GIP release seems to be dependent on the rate of absorption rather than the mere presence of nutrients [24, 25]. A number of studies have shown that all three macronutrients (carbohydrate, protein, and fat) are capable of stimulating the release of the incretin hormones (reviewed in [22, 26]), although there does seem to be some difference in the response patterns of each of these three nutrients. Carbohydrates are a good stimulus for both GLP-1 and GIP secretion [19, 21]. However, while the GLP-1 response is similar after ingestion of isocaloric fat or protein loads, protein appears to be a more potent stimulus than fat for the early release of GIP [27].
The precise mechanism by which macronutrients cause release of the incretin hormones is not fully understood, but it is believed that direct interaction between the ingested nutrients and the K- and L-cells may be the principal mechanism involved (Figure 1). In vitro studies have demonstrated that changes in membrane potential and mobilization of intracellular calcium following glucose stimulation lead to GLP-1 and GIP release [28, 29]. Several glucose-sensing pathways have been suggested to be involved, including active glucose uptake via the sodium-glucose cotransporter SGLT-1 [29-31], and closure of ATP-sensitive K+ channels [29, 32]. More recently, a mechanism involving activation of the sweet taste-receptor, gustducin, has also been proposed [33]. However, it is still unclear whether all of the above mentioned pathways are involved in mediating glucose-induced incretin hormone secretion in humans, since neither sulphonylureas nor ingestion of non-calorific non-metabolizable sweeteners have been observed to affect incretin levels in clinical studies [34-36]. It has been thought that glucose was able to stimulate GLP-1 secretion only via interaction with the apical (luminal) membrane of the L-cell, as intravenously infused glucose was not thought to result in elevated GLP-1 levels. However, GLP-1 secretion from the isolated perfused intestine was stimulated by high glucose concentrations in the perfusate, suggesting that glucose in the blood may be capable of stimulating GLP-1 release as well [37]. This suggestion has been supported by the development of more sensitive GLP-1 assays. Using these assays, it has been possible to demonstrate that hyperglycemia itself can lead to small, but significant, increases in GLP-1 levels [38]. However, the mechanism by which circulating glucose stimulates the L-cells remains unknown.
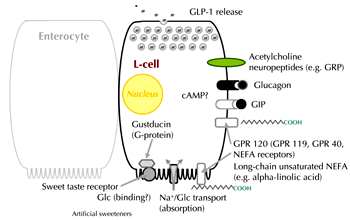 |
 |
Figure 1. Proposed mechanisms involved in stimulating L-cell secretion. Absorption of nutrients over the luminal (brush border) membrane may be the predominant mechanism in humans, although other signals may include neurotransmitters, hormones and bile acids (see text for details). cAMP: cyclic adenosine monophosphate. GIP: glucose-dependent insulinotropic polypeptide. Glc: glucose. GLP-1: glucagon-like peptide-1. GRP: G-protein-coupled receptor. Na: sodium. NEFA: non-esterified fatty acid. Reproduced with kind permission from [122] (Springer Science+Business Media; Diabetologia, 2011, 54:10-18, Nauck et al., Fig. 1). |
|
The mechanisms mediating the effects of other macronutrients on GLP-1 and GIP secretion have not been well characterized. Amino acids such as alanine, glutamine, and asparagine can stimulate GLP-1 secretion via mechanisms which may involve activation of ligand-gated ion channels or sodium-coupled uptake. Both pathways lead to membrane depolarization, calcium influx, and finally GLP-1 release [39-41]. Enteroendocrine cells express a number of different G-protein-coupled receptors (GPR) [31, 42], some of which have been proposed to mediate the stimulatory effects of dietary fats on incretin hormones. Naturally occurring lipid amides, such as oleoylethanolamide, and 2-monoacylglycerols, such as 2-oleoyl glycerol, have been shown to act as endogenous ligands of GPR119 [43, 44]. Activation of GPR119 is associated with increased cyclic AMP levels and GLP-1 release in mice [45] and humans [44]. Other GPR have been identified which are responsive to long-chain fatty acids, (e.g. GPR40, GPR120) and which have been suggested to mediate free fatty acid stimulation of GLP-1 [46, 47].
Lipids may also indirectly stimulate GLP-1 secretion via bile acids. Thus, activation of the bile acid receptor TGR5 promotes GLP-1 secretion from an enteroendocrine cell line in vitro [48], and GLP-1 release from the perfused rat colon increases following perfusion with bile acids [49]. Furthermore, preliminary data have shown that intra-rectal infusion of bile acids is associated with increased GLP-1 levels in patients with type 2 diabetes [50]. However, the precise physiological importance of this pathway in humans is unclear, with another preliminary report indicating that postprandial GLP-1 responses are not impaired in cholecystectomized patients [51].
While both K- and L-cells undoubtedly respond to direct stimulation, it has been argued that other mechanisms must be involved to explain the early rise in incretin levels, particularly for GLP-1, where plasma concentrations increase before nutrients reach the more distally located L-cells.
Neural regulation
A number of studies suggest that incretin hormone secretion can be influenced by autonomic nerves, with in vitro and animal studies indicating that vagal cholinergic muscarinic pathways are involved [26]. In rodents, a transient increase in GLP-1 levels was observed in anticipation of food being presented [52], suggesting a cephalic response. However, in humans, the balance of evidence suggests that vagal cholinergic innervation does not play a major role in regulating incretin hormone secretion. Sham-feeding does not alter GLP-1 concentrations [53], while basal GLP-1 and GIP concentrations remain unchanged in the pre-absorptive (cephalic) period [54], and are unaffected by the non-specific muscarinic blocker atropine. Furthermore, there are no major changes in prandial responses following small intestinal resection [55]. A role for the sympathetic nervous system and for non-adrenergic non-cholinergic neurons has been suggested from animal studies. However, it remains unclear whether these pathways play a significant role in the regulation of incretin hormone secretion in humans [26].
Hormonal regulation
An endocrine loop linking the duodenum to the ileum has also been proposed as a mechanism to explain the paradoxically early rise in GLP-1 levels. GIP, in particular, was shown to be capable of increasing GLP-1 release in a several animal studies, but a role for hormones in stimulating the secretion of GLP-1 in humans has not been established [26]. However, there is some evidence to support the notion that local paracrine mechanisms may act to restrain incretin hormone secretion. It has been observed that endogenous GLP-1 levels are suppressed following infusion of exogenous GLP-1 in humans [56]. Furthermore, DPP-4 inhibition, which leads to increased levels of endogenous intact (active) GLP-1 and GIP, is associated with lower total incretin levels, in both animals [57] and humans [34], suggesting that their secretion has been suppressed. These observations support the suggestion that there may be a local feedback loop, whereby the increased levels of intact incretin hormones restrain further secretion [57]. It has been proposed that this is mediated by somatostatin from neighboring δ-cells exerting a paracrine inhibitory effect [58].
The most important stimulus for incretin hormone release in humans is probably the direct stimulation of K- and L-cells, with the explanation for the rapid early increase in GLP-1 level following food ingestion likely to be due to the stimulation of the proximally located L-cells. Notably, surgical removal of the distal intestine has not been observed to affect the GLP-1 response to glucose [55]. It has been shown that nutrients first reach the proximal duodenum within 6-8 minutes following ingestion of a liquid glucose load, which correlates well with the initial increase in GLP-1 levels [24]. Calculation of the amount of GLP-1 present in the upper intestine indicates that this is more than sufficient to account for the GLP-1 response to oral glucose [59]. The extent of the GLP-1 response is related to both meal composition and meal size [60]. Moreover, there is also a diurnal variation, with responses being greater when an identical meal is served in the morning compared to the afternoon [61].
Incretin hormone action
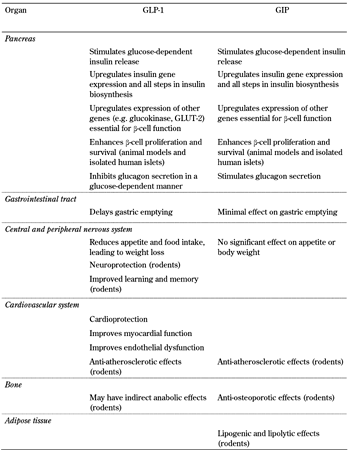
The role conventionally assigned to incretin hormones is that of enhancing glucose-stimulated insulin secretion. However, both GLP-1 and GIP have multiple actions which influence blood glucose levels beyond simply stimulating insulin secretion. Moreover, it is becoming increasingly clear that they exert a number of other effects in addition to those involved in the regulation of glucose homeostasis (Table 1).
Table
1.
Actions of GLP-1 and GIP in the different organs |
|
 |
|
The actions of GLP-1 are mediated by specific GPR found in its target organs such as the pancreas, brain, heart, kidney, and lung, but the presence of the GLP-1 receptor in other tissues such as liver, muscle, and fat cells remains controversial [62]. Many of the peptide's effects in these other tissues are likely to be indirect, at least in humans. GLP-1 has potent effects on pancreatic β-cells; it enhances the meal-stimulated release of insulin (the "incretin effect") in a glucose-dependent manner, stimulates all stages of insulin biosynthesis, and improves overall β-cell function [62]. Moreover, animal and in vitro studies have demonstrated that it has a β-cell protective effect, promoting differentiation and proliferation, while reducing apoptosis, which results in increased β-cell mass [62]. However, effects on β-cell mass have not yet been demonstrated in humans. GLP-1 also potently suppresses α-cell secretion of glucagon, again in a glucose-dependent manner. However, reports of GLP-1 receptor expression on α-cells have been inconsistent [63, 64], and the glucagonostatic effect of GLP-1 is believed to be an indirect effect, mediated by insulin, zinc, or GABA released from the β-cells [65, 66], and/or somatostatin released from the neighboring δ-cells [67]. Together, the insulinotropic and glucagonostatic effects of GLP-1 have been shown to exert a powerful effect on blood glucose levels. Studies using the GLP-1 receptor antagonist exendin 9-39 have revealed that endogenous GLP-1 is important for the regulation of normal glucose homeostasis. Thus, when infused into healthy humans, exendin 9-39 was associated with increased postprandial glucose excursions [68]. Since this finding, several clinical studies have demonstrated that endogenous GLP-1 plays a major role in regulating pancreatic islet responses to glucose [69, 70]. When GLP-1 action is blocked glucose-induced insulin secretion is lower and the incretin effect is reduced by around 50% [69]. Both the stimulation of insulin and the suppression of glucagon secretion are important mediators, and have been demonstrated to contribute almost equally to the glucose-lowering effect of GLP-1 [71].
GLP-1 also reduces gastric emptying, which has been proposed to be the principal mechanism by which GLP-1 reduces postprandial glucose excursions [72]. Nevertheless, it remains unclear how much this mechanism contributes to the glucose-lowering effect of endogenous GLP-1, since GLP-1 receptor antagonism using exendin 9-39 affected gastric empting in some [73], but not all [70, 74], studies.
Numerous studies have indicated that GLP affects appetite and food intake in humans [75], with both peripheral and central mechanisms being proposed as mediators [62]. Exogenous GLP-1 and GLP-1 receptor agonists used clinically have consistently been associated with body weight loss, whereas enhancement of endogenous GLP-1 levels using DPP-4 inhibitors is generally body weight neutral [75, 76]. This may suggest that higher GLP-1 concentrations are necessary to elicit the body weight-lowering effect than are required for the antihyperglycemic effects. However, the extent to which GLP-1 plays a physiological role in regulating appetite/satiety and body weight is unclear. In rodent studies, investigating the role of GLP-1 on food intake, GLP-1 receptor antagonism using exendin 9-39 has given conflicting results [77-79].
GLP-1 receptors are also found throughout the cardiovascular system, and data are accumulating from both animal and clinical studies which suggest that both exogenous and endogenous GLP-1 may be cardioprotective and have beneficial cardiovascular actions. These actions are reviewed elsewhere in this issue [80, 81]. There is also preclinical evidence that GLP-1 may exert neuroprotective effects. GLP-1 receptors have been demonstrated on both peripheral and central neurons, and favorable effects of GLP-1 have been observed in rodent models of Parkinsonism and Alzheimer's disease (reviewed in [82]). Moreover, recent data from animal studies suggest that GLP-1 may have the potential to ameliorate peripheral neuropathy. GLP-1 receptor activation was reported to be associated with both histological and functional improvements in a rat model of pyridoxine-induced peripheral sensory neuropathy [83]. Diabetes-induced peripheral nerve degeneration was reduced in studies using a GLP-1 receptor agonist or a DPP-4 inhibitor to enhance endogenous GLP-1 levels [84, 85], while hyperglycemia-induced reductions of motor nerve conduction velocity were attenuated by GLP-1 receptor agonism in diabetic mice [86]. However, as yet, no studies have addressed potential neuroprotective properties of GLP-1 in humans.
Like the GLP-1 receptor, the GIP receptor is also a GPR, found in numerous tissues, including pancreas, bone, brain, and adipocytes [62]. GIP shares many of the GLP-1 effects on the β-cell, where it increases insulin biosynthesis and secretion and exerts a β-cell protective effect [62]. However, its effect on the α-cell contrasts with that of GLP-1; GIP stimulates the release of glucagon from the perfused rat pancreas [67], and has glucagonotropic actions in humans [87]. GIP has been implicated in lipid metabolism and adiposity from studies in rodents [62]. Knock-out of the GIP receptor confers resistance to diet-induced obesity in mice [88], although there is no clear evidence at present to suggest an association between GIP and obesity in humans. In bone, GIP receptors are located on both osteoclasts and osteoblasts, and studies in mice and rats have indicated that GIP signaling is involved in the regulation of bone turnover. Bone formation is enhanced while bone resorption is suppressed, which leads to increases in bone mass and bone mineral density [89]. However, although the GIP receptor has been demonstrated in some human bone cell-lines [90], acute administration of GIP has not been associated with changes in markers of bone resorption in humans [91].
Incretin hormones in disease states
Interpretation of the precise relationship between disease and incretin hormone secretion and the incretin effect is complicated. However, there is evidence to suggest that impairments in secretion and/or action of incretin hormones arise secondarily to the development of insulin resistance, glucose intolerance, and/or increases in body weight rather than being causative factors. In separate studies, insulin sensitivity, glucose tolerance, and body mass index (BMI) have all been identified as independent factors associated with reductions in GLP-1 secretion and an impaired incretin effect [92-94].
Obesity
The incretin effect has been reported to be reduced in obese subjects with normal glucose tolerance [94, 95], but it is unclear whether this is due to reduced secretion of incretin hormones and/or impairments in their action. After mixed meal and glucose ingestion, the secretion of GLP-1, but not of GIP, is reduced in obese subjects compared to lean subjects [27]. There is also evidence for a link between body weight and GLP-1 from a number of studies, all showing an inverse relationship between body weight and GLP-1 levels [27, 93, 94, 96]. Meal-stimulated GLP-1 levels are impaired in obese subjects with normal glucose tolerance compared with lean subjects, with some partial recovery in levels following weight loss [97]. However, there is little consensus regarding the relationship between obesity and GIP secretion. Most studies have found no impact of changes in body weight on GIP levels [27, 94, 97], although in other studies, both modestly reduced [93] and increased [98] GIP levels have been reported in obese subjects.
At present, only a few studies have addressed the subject of whether incretin hormone action is altered by obesity. One study has shown that the insulinotropic ability of GIP is similar in obese and lean subjects [99]. In contrast, an animal study demonstrated an augmented insulinotropic action of GLP-1 in high-fat fed obese and insulin-resistant mice compared with normal mice [100].
Subjects at risk of developing type 2 diabetes
Normal glucose-tolerant subjects at increased risk of subsequently developing diabetes have been studied to find out whether impaired incretin secretion and/or action is apparent before overt diabetes appears. Incretin hormone concentrations appear normal in non-diabetic offspring of type 2 diabetes patients [101] and in first-degree relatives of diabetic subjects [102], whereas, in identical twins discordant for diabetes, reduced GLP-1 levels were observed in the affected twin only [103]. In women with gestational diabetes, impaired GLP-1 responses have been reported [104], but these return to normal following birth and re-establishment of normal glucose tolerance [104, 105].
Data are conflicting regarding alterations in incretin hormones in subjects with impaired glucose tolerance. Normal GLP-1 responses were reported in some subjects with impaired glucose tolerance [96, 106], whereas an impairment was identified in others. However, further analysis concluded that the reductions could largely be explained by differences in BMI and/or insulin sensitivity [93, 107]. In terms of incretin action in impaired glucose-tolerant subjects, the insulinotropic effect of GLP-1 was found to be reduced [108], and an impaired incretin effect has been reported [107]. In another study, GIP action was normal in subjects with previous gestational diabetes [105].
Type 2 diabetes
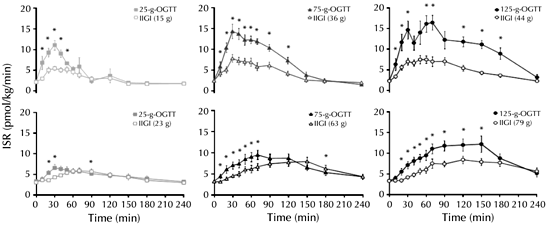
In patients with type 2 diabetes, the incretin effect is clearly reduced [109, 110], which results in an inappropriately low insulin response to the ingestion of nutrients (Figure 2). Several early studies indicated that the reduced incretin effect could, at least in part, be related to impaired secretion of GLP-1 (whereas secretion of GIP is generally found to be unaltered). Impaired meal-stimulated GLP-1 levels have been reported in some studies of patients with type 2 diabetes [93, 94, 111]. However, in other studies, no such impairment was observed [96, 112]. Moreover, it is now being recognized that some antidiabetic agents themselves are able to influence incretin hormone levels. This finding further complicates the interpretation of data from different studies in which incretin levels were assessed. In this regard, the different treatment regimens are important. In some studies, patients were still on drug therapy whereas in others, therapy had been discontinued and patients were studied after washout periods of differing duration.
 |
|
Figure 2. Impaired regulation of the incretin effect in patients with type 2 diabetes. Insulin secretion rates (ISR) in healthy control subjects (upper panel) and patients with type 2 diabetes (lower panel) following low (25 g), medium (75 g), and high (125 g) oral glucose loads (closed symbols) and corresponding isoglycemic intravenous glucose infusions (open symbols). Figures in brackets indicate the amount of glucose infused to mimic the glycemic excursion following the oral glucose. * p < 0.05. IIGI: isoglycemic intravenous glucose infusion. ISR: insulin secretion rate. OGTT: oral glucose tolerance test. Reproduced with kind permission from [110] (The Endocrine Society, J Clin Endocrinol Metab, 2011, 96:373-345, Bagger et al., Fig. 2c). |
|
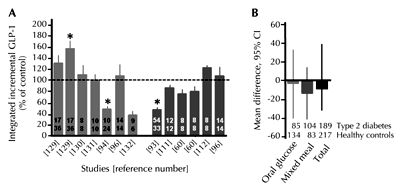
It is now well established that DPP-4 inhibitors increase intact incretin hormone concentrations [113]. However, metformin is also associated with increased GLP-1 responses to food ingestion in animals [114] and humans [115], at least in acute studies, although the mechanism has not been fully elucidated. Metformin is not a competitive inhibitor of DPP-4, but results from preclinical studies have suggested that it may upregulate proglucagon expression in the intestinal L-cells [115]. Furthermore, preliminary data have indicated that metformin may also influence GLP-1 levels indirectly, mediated via increased bile acid levels. In mice, metformin was reported to inhibit the apical sodium-dependent bile acid transporter (ASBT), resulting in increased bile acid concentrations in the intestine and increased plasma GLP-1 levels [116]. In accordance with this finding, it has recently been reported that bile acid sequestrants such as colesevelam have been associated with increased plasma GLP-1 concentrations [117, 118]. Other studies have demonstrated that α-glucosidase inhibitors such as acarbose and miglitol also stimulate GLP-1 secretion in humans [119, 120]. Again, the mechanism is unclear, but may involve increased exposure of the L-cells to nutrients. On the other hand, it has also been reported that in mice, α-glucosidase inhibition is associated with elevated proglucagon gene expression and GLP-1 content in the intestine [121]. Thus, while it appears that some patients with type 2 diabetes are characterized by a reduction in meal-related GLP-1 responses, this may not be a universal trait common to all patients. In particular those with well controlled glycemia may have relatively normal GLP-1 levels (Figure 3) [122].
 |
|
Figure 3. GLP-1 response to glucose. A: Integrated "total" GLP-1 response to oral glucose (light grey bars) or mixed meal (dark grey bars) in type 2 diabetic patients, expressed as percentage of the response in matched non-diabetic control subjects. Figures in the bars refer to the number of type 2 diabetic patients (upper row) or control subjects (lower row) studied. Figures below the bars refer to the original studies. B: Meta-analysis of published studies reporting GLP-1 responses in healthy individuals and subjects with type 2 diabetes after a 75g oral glucose tolerance test and/or a standardized mixed meal. GLP-1: glucagon-like peptide-1. CI: confidence interval. Modified and reproduced with kind permission from [122] (Springer Science+Business Media, Diabetologia, 2011, 54:10-18, Nauck et al., Fig. 2b). |
|
There does seem to be general agreement that the actions of the incretin hormones are impaired in type 2 diabetes. Although GLP-1 retains its insulinotropic effect in patients with type 2 diabetes, its potency is reduced [123, 124]. In contrast, the actions of GIP are severely impaired [124-126], with an almost complete loss of amplification of the second-phase insulin response. However, it seems unlikely that the impaired incretin effect itself is the cause of type 2 diabetes. Rather, it would appear to develop subsequently to insulin resistance, bodyweight, and/or glucose intolerance, which may contribute to the deterioration of glycemic control in diabetic patients. In accordance with this consideration, similar impairments in incretin actions are seen in diabetic patients irrespective of etiology or phenotype [127]. Moreover, in healthy subjects, an impaired incretin effect has been reported, following short-term (12 days) glucocorticoid treatment to induce reduced glucose tolerance and insulin resistance [128]. Conversely, strict glycemic control to bring about near-normalization of blood glucose levels in patients with type 2 diabetes leads to improvements (although not normalization) in the ability of GLP-1 and GIP to enhance insulin secretion [124].
Conclusions
Incretin hormone secretion is regulated by a variety of different signaling pathways, which allow a coordinated response to a physiological stimulus. Many factors (nutrients, nerves, hormones, and even drugs) can affect K- and L-cell responses via direct and indirect mechanisms. However in humans, direct stimulation by nutrients in contact with the apical membrane of the endocrine cells appears to be the principal factor determining incretin hormone secretion.
Alterations in GLP-1 and GIP responses are reported to be related to changes in body weight, insulin resistance, and glucose tolerance, but the precise ways in which these factors impact on the enteroendocrine cells remains unclear. Nevertheless, changes in incretin hormone levels have been reported in obese glucose-tolerant subjects, with impairments in incretin action becoming evident as glycemic control deteriorates. These impairments probably contribute to the ongoing reduction in β-cell function and developing hyperglycemia observed when overt diabetes becomes established.
Emerging data are suggesting that incretin hormones also have other actions in addition to their accepted roles in the regulation of normal islet function and glucose homeostasis. These include potentially beneficial cardiovascular and neuroprotective actions. However, while the glucoregulatory actions of the incretins have been convincingly demonstrated to be impaired in subjects with diabetes, it is currently unknown whether this also applies to any of their non-glucose effects. Given the increasing use of incretin-based therapies as part of the treatment paradigm for type 2 diabetes, further investigation of the pleiotropic effects of the incretin hormones in disease states is clearly warranted.
Disclosures: The authors report no conflict of interests.
References
- Creutzfeldt W. The incretin concept today. Diabetologia 1979. 16:75-85. [DOD] [CrossRef]
- Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, Creutzfeldt W. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab 1986. 63:492-498. [DOD] [CrossRef]
- Lindgren O, Carr RD, Deacon CF, Holst JJ, Pacini G, Mari A, Ahren B. Incretin hormone and insulin responses to oral versus intravenous lipid administration in humans. J Clin Endocrinol Metab 2011. 96:2519-2524. [DOD] [CrossRef]
- Eissele R, Göke R, Willemer S, Harthus HP, Vermeer H, Arnold R, Göke B. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur J Clin Invest 1992. 22:283-291. [DOD] [CrossRef]
- Mortensen K, Christensen LL, Holst JJ, Orskov C. GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept 2003. 114:189-196. [DOD] [CrossRef]
- Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 1995. 80:952-957. [DOD] [CrossRef]
- Deacon CF, Nauck MA, Meier J, Hucking K, Holst JJ. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab 2000. 85:3575-3581. [DOD] [CrossRef]
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995. 44:1126-1131. [DOD] [CrossRef]
- Holst JJ, Deacon CF. Inhibition of the activity of dipeptidyl-peptidase IV as a treatment for type 2 diabetes. Diabetes 1998. 47:1663-1670. [DOD] [CrossRef]
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007. 87:1409-1439. [DOD] [CrossRef]
- Heller RS, Aponte GW. Intra-islet regulation of hormone secretion by glucagon-like peptide-1-(7-36) amide. Am J Physiol 1995. 269:G852-G860. [DOD]
- Whalley N, Pritchard L, Smith DM, White A. Processing of proglucagon to GLP-1 in pancreatic alpha cells: is this a paracrine mechanism enabling GLP-1 to act on beta cells? J Endocrinol 2011. In press. [DOD]
- Orskov C, Rabenhoj L, Wettergren A, Kofod H, Holst JJ. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 1994. 43:535-539. [DOD] [CrossRef]
- Ugleholdt R, Poulsen ML, Holst PJ, Irminger JC, Orskov C, Pedersen J, Rosenkilde MM, Zhu X, Steiner DF, Holst JJ. Prohormone convertase 1/3 is essential for processing of the glucose-dependent insulinotropic polypeptide precursor. J Biol Chem 2006. 281:11050-11057. [DOD] [CrossRef]
- Fujita Y, Wideman RD, Asadi A, Yang GK, Baker R, Webber T, Zhang T, Wang R, Ao Z, Warnock GL, Kwok YN, Kieffer TJ. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology 2010. 138:1966-1975. [DOD] [CrossRef]
- Deacon CF, Pridal L, Klarskov L, Olesen M, Holst JJ. Glucagon-like peptide 1 undergoes differential tissue-specific metabolism in the anesthetized pig. Am J Physiol 1996. 271:E458-E464. [DOD]
- Vilsboll T, Agerso H, Krarup T, Holst JJ. Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. J Clin Endocrinol Metab 2003. 88:220-224. [DOD] [CrossRef]
- Vilsboll T, Agerso H, Lauritsen T, Deacon CF, Aaboe K, Madsbad S, Krarup T, Holst JJ. The elimination rates of intact GIP as well as its primary metabolite, GIP 3-42, are similar in type 2 diabetic patients and healthy subjects. Regul Pept 2006. 137:168-172. [DOD] [CrossRef]
- Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V. Glucagon-like peptide-1 (7-36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24-h secretion patterns. J Endocrinol 1993. 138:159-166. [DOD] [CrossRef]
- Orskov C, Wettergren A, Holst JJ. Secretion of the incretin hormones glucagon-like peptide-1 and gastric inhibitory polypeptide correlates with insulin secretion in normal man throughout the day. Scand J Gastroenterol 1996. 31:665-670. [DOD] [CrossRef]
- Carr RD, Larsen MO, Winzell MS, Jelic K, Lindgren O, Deacon CF, Ahren B. Incretin and islet hormonal responses to fat and protein ingestion in healthy men. Am J Physiol Endocrinol Metab 2008. 295:E779-E784. [DOD] [CrossRef]
- Ahren B, Carr RD, Deacon CF. Incretin hormone secretion over the day. Vitam Horm 2010. 84:203-220. [DOD] [CrossRef]
- Miholic J, Orskov C, Holst JJ, Kotzerke J, Meyer HJ. Emptying of the gastric substitute, glucagon-like peptide-1 (GLP-1), and reactive hypoglycemia after total gastrectomy. Dig Dis Sci 1991. 36:1361-1370. [DOD] [CrossRef]
- Schirra J, Katschinski M, Weidmann C, Schäfer T, Wank U, Arnold R, Göke B. Gastric emptying and release of incretin hormones after glucose ingestion in humans. J Clin Invest 1996. 97:92-103. [DOD] [CrossRef]
- Herrmann C, Göke R, Richter G, Fehmann HC, Arnold R, Göke B. Glucagon-like peptide-1 and glucose-dependent insulin-releasing polypeptide plasma levels in response to nutrients. Digestion 1995. 56:117-126. [DOD] [CrossRef]
- Deacon CF. What do we know about the secretion and degradation of incretin hormones? Regul Pept 2005. 128:117-124. [DOD]
- Carr RD, Larsen MO, Jelic K, Lindgren O, Vikman J, Holst JJ, Deacon CF, Ahren B. Secretion and dipeptidyl peptidase-4-mediated metabolism of incretin hormones after a mixed meal or glucose ingestion in obese compared to lean, nondiabetic men. J Clin Endocrinol Metab 2010. 95:872-878. [DOD] [CrossRef]
- Reimann F, Gribble FM. Glucose-sensing in glucagon-like peptide-1-secreting cells. Diabetes 2002. 51:2757-2763. [DOD] [CrossRef]
- Gribble FM, Williams L, Simpson AK, Reimann F. A novel glucosesensing mechanism contributing to glucagon-like peptide-1 secretion from the GLUTag cell line. Diabetes 2003. 52:1147-1154. [DOD] [CrossRef]
- Sykes S, Morgan LM, English J, Marks V. Evidence for preferential stimulation of gastric inhibitory polypeptide secretion in the rat by actively transported carbohydrates and their analogues. J Endocrinol 1980. 85: 201-207. [DOD] [CrossRef]
- Parker HE, Habib AM, Rogers GJ, Gribble FM, Reiman F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 2009. 52:289-298. [DOD] [CrossRef]
- Wang SY, Chi MM, Li L, Moley KH, Wice BM. Studies with GIP/Ins cells indicate secretion by gut K cells is KATP channel independent. Am J Physiol Endocrinol Metab 2003. 284:E988-E1000. [DOD]
- Jang HJ, Kokrashvili Z, Theodorakis MJ, Carlson OD, Kim BJ, Zhou J, Kim HH, Xu X, Chan SL, Juhaszova M, Bernier M, Mosinger B, Margolskee RF, Egan JM. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci USA 2007. 104:15069-15074. [DOD] [CrossRef]
- El-Ouaghlidi A, Rehring E, Holst JJ, Schweizer A, Foley J, Holmes D, Nauck MA. The dipeptidyl peptidase 4 inhibitor vildagliptin does not accentuate glibenclamide-induced hypoglycemia but reduces glucose-induced glucagon-like peptide 1 and gastric inhibitory polypeptide secretion. J Clin Endocrinol Metab 2007. 92:4165-4171. [DOD] [CrossRef]
- Ma J, Bellon M, Wishart JM, Young R, Blackshaw LA, Jones KL, Horowitz M, Rayner CK. Effect of the artificial sweetener, sucralose, on gastric emptying and incretin hormone release in healthy subjects. Am J Physiol Gastrointest Liver Physiol 2009. 296:G735-G739. [DOD] [CrossRef]
- Ford HE, Peters V, Martin NM, Sleeth ML, Ghatei MA, Frost GS, Bloom SR. Effects of oral ingestion of sucralose on gut hormone response and appetite in healthy normal-weight subjects. Eur J Clin Nutr 2011. 65:508-513. [DOD] [CrossRef]
- Hansen L, Hartmann B, Mineo H, Holst JJ. Glucagon-like peptide-1 secretion is influenced by perfusate glucose concentration and by a feedback mechanism involving somatostatin in isolated perfused porcine ileum. Regul Pept 2004. 118:11-18. [DOD] [CrossRef]
- Vardarli I, Nauck MA, Köthe LD, Deacon CF, Holst JJ, Schweizer A, Foley JE. Inhibition of DPP-4 with vildagliptin improved insulin secretion in response to oral as well as "isoglycemic" intravenous glucose without numerically changing the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab 2011. 96:945-954. [DOD] [CrossRef]
- Reimann F, Williams L, da Silva Xavier G, Rutter GA, Gribble FM. Glutamine potently stimulates glucagon-like peptide-1 secretion from GLUTag cells. Diabetologia 2004. 47:1592-1601. [DOD] [CrossRef]
- Gameiro A, Reimann F, Habib AM, O'Malley D, Williams L, Simpson AK, Gribble FM. The neurotransmitters glycine and GABA stimulate glucagon-like peptide-1 release from the GLUTag cell line. J Physiol 2005. 569:761-772. [DOD] [CrossRef]
- Tolhurst G, Zheng Y, Parker HE, Habib AM, Reimann F, Gribble FM. Glutamine triggers and potentiates glucagon-like peptide-1 secretion by raising cytosolic Ca2+ and cAMP. Endocrinology 2011. 152:405-413. [DOD] [CrossRef]
- Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose sensing in L cells: a primary cell study. Cell Metab 2008. 8:532-539. [DOD] [CrossRef]
- Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab 2006. 3:167-175. [DOD] [CrossRef]
- Hansen KB, Rosenkilde MM, Knop FK, Wellner N, Diep TA, Rehfeld JF, Andersen UB, Holst JJ, Hansen HS. 2-oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J Clin Endocrinol Metab 2011. In press. [DOD]
- Chu ZL, Carroll C, Alfonso J, Gutierrez V, He H, Lucman A, Pedraza M, Mondala H, Gao H, Bagnol D, Chen R, Jones RM, Behan DP, Leonard J. A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like peptide-1 and glucose-dependent insulinotropic peptide release. Endocrinology 2008. 149:2038-2047. [DOD] [CrossRef]
- Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 2005. 11:90-94. [DOD] [CrossRef]
- Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008. 57:2280-2287. [DOD] [CrossRef]
- Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun 2005. 329:386-390. [DOD] [CrossRef]
- Plaisancie P, Dumoulin V, Chayvialle JA, Cuber JC. Luminal glucagon-like peptide-1(7-36) amide-releasing factors in the isolated vascularly perfused rat colon. J Endocrinol 1995. 145:521-526. [DOD] [CrossRef]
- Adrian TE, Gariballa S, Parekh KA, Thomas SA, Saadi H, Al Kaabi J, Nagelkerke N, Gedulin B, Young AA. The bile acid brake: a novel target for treating diabetes and obesity. Diabetes 2010. 59(Suppl 1):A164. [DOD]
- Sonne D, Hare KJ, Martens PC, Hansen KM, Holst JJ, Vilsboll T, Knop FK. Preserved postprandial GLP-1 responses in cholecystectomized subjects: No evidence of a physiological role of gallbladder emptying in postprandial GLP-1 release. 71st Scientific Sessions of the American Diabetes Association 2011. Abstract 107-LB). Available at http://ww2.aievolution.com/ada1101/index.cfm?do=abs.viewAbs&abs=12080. [DOD]
- Vahl TP, Drazen DL, Seeley RJ, D'Alessio DA, Woods SC. Meal-anticipatory glucagon-like peptide-1 secretion in rats. Endocrinology 2010. 151:569-575. [DOD] [CrossRef]
- Wettergren A, Petersen H, Orskov C, Christiansen J, Sheikh SP, Holst JJ. Glucagon-like peptide-1 7-36 amide and peptide YY from the L-cell of the ileal mucosa are potent inhibitors of vagally induced gastric acid secretion in man. Scand J Gastroenterol 1994. 29:501-505. [DOD] [CrossRef]
- Ahren B, Holst JJ. The cephalic insulin response to meal ingestion in humans is dependent on both cholinergic and noncholinergic mechanisms and is important for postprandial glycemia. Diabetes 2001. 50:1030-1038. [DOD] [CrossRef]
- Nauck MA, Siemsglüss J, Orskov C, Holst JJ. Release of glucagon-like peptide 1 (GLP-1 [7-36 amide]), gastric inhibitory polypeptide (GIP) and insulin in response to oral glucose after upper and lower intestinal resections. Z Gastroenterol 1996. 34:159-166. [DOD]
- Toft-Nielsen MB, Madsbad S, Holst JJ. Continuous subcutaneous infusion of glucagon-like peptide 1 lowers plasma glucose and reduces appetite in type 2 diabetic patients. Diabetes Care 1999. 22:1137-1143. [DOD] [CrossRef]
- Deacon CF, Wamberg S, Bie P, Hughes TE, Holst JJ. Preservation of active incretin hormones by inhibition of dipeptidyl peptidase IV suppresses meal-induced incretin secretion in dogs. J Endocrinol 2002. 172:355-362. [DOD] [CrossRef]
- Hansen L, Hartmann B, Bisgaard T, Mineo H, Jorgensen PN, Holst JJ. Somatostatin restrains the secretion of glucagon-like peptide-1 and -2 from isolated perfused porcine ileum. Am J Physiol Endocrinol Metab 2000. 278:E1010-E1018. [DOD]
- Nauck MA. Glucagon-like peptide 1 (GLP-1): a potent gut hormone with a possible therapeutic perspective. Acta Diabetol 1998. 35: 117-129. [DOD] [CrossRef]
- Vilsboll T, Krarup T, Sonne J, Madsbad S, Volund A, Juul AG, Holst JJ. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab 2003. 88:2706-2713. [DOD] [CrossRef]
- Lindgren O, Mari A, Deacon CF, Carr RD, Winzell MS, Vikman J, Ahren B. Differential islet and incretin hormone responses in morning versus afternoon after standardized meal in healthy men. J Clin Endocrinol Metab 2009. 94:2887-2892. [DOD] [CrossRef]
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007. 132:2131-2157. [DOD] [CrossRef]
- Moens K, Heimberg H, Flamez D, Huypens P, Quartier E, Ling Z, Pipeleers D, Gremlich S, Thorens B, Schuit F. Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 1996. 45:257-261. [DOD] [CrossRef]
- Heller RS, Kieffer TJ, Habener JF. Insulinotropic glucagon like peptide I receptor expression in glucagon-producing alpha cells of the rat endocrine pancreas. Diabetes 1997. 46:785-791. [DOD] [CrossRef]
- Franklin J, Gromada J, Glinovci A, Theander S, Wollheim CB. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005. 54:1808-1815. [DOD] [CrossRef]
- Bailey SJ, Ravier MA, Rutter GA. Glucose-dependent regulation of gamma-aminobutyruc acid (GABA A) receptor expression in mouse pancreaytic islet alpha-cells. Diabetes 2007. 56:320-327. [DOD] [CrossRef]
- de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 2008. 51:2263-2270. [DOD] [CrossRef]
- Edwards CM, Todd JF, Mahmoudi M, Wang Z, Wang RM, Ghatei MA, Bloom SR. Glucagon-like peptide 1 has a physiological role in the control of postprandial glucose in humans: studies with the antagonist exendin 9-39. Diabetes 1999. 48:86-93. [DOD] [CrossRef]
- Schirra J, Nicolaus M, Roggel R, Katschinski M, Storr M, Woerle HJ, Göke B. Endogenous glucagon-like peptide 1 controls endocrine pancreatic secretion and antro-pyloro-duodenal motility in humans. Gut 2006. 55:243-251. [DOD] [CrossRef]
- Salehi M, Vahl TP, D'Alessio DA. Regulation of islet hormone release and gastric emptying by endogenous glucagon-like peptide 1 after glucose ingestion. J Clin Endocrinol Metab 2008. 93:4909-4916. [DOD] [CrossRef]
- Hare KJ, Vilsboll T, Asmar M, Deacon CF, Knop FK, Holst JJ. The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes 2010. 59:1765-1770. [DOD] [CrossRef]
- Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, Schmiegel WH. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol 1997. 273:E981-E988. [DOD]
- Deane AM, Nguyen NQ, Stevens JE, Fraser RJ, Holloway RH, Besanko LK, Burgstad C, Jones KL, Chapman MJ, Rayner CK, Horowitz M. Endogenous glucagon-like peptide-1 slows gastric emptying in healthy subjects, attenuating postprandial glycemia. J Clin Endocrinol Metab 2010. 95:215-221. [DOD] [CrossRef]
- Nicolaus M, Brödl J, Linke R, Woerle HJ, Göke B, Schirra J. Endogenous GLP-1 regulates postprandial glycemia in humans: relative contributions of insulin, glucagon, and gastric emptying. J Clin Endocrinol Metab 2011. 96:229-236. [DOD] [CrossRef]
- Verdich C, Flint A, Gutzwiller JP, Näslund E, Beglinger C, Hellström PM, Long SJ, Morgan LM, Holst JJ, Astrup A. A meta-analysis of the effect of glucagon-like peptide-1 (7-36) amide on ad libitum energy intake in humans. J Clin Endocrinol Metab 2001. 86:4382-4389. [DOD] [CrossRef]
- Madsbad S, Krarup T, Deacon CF, Holst JJ. Glucagon-like peptide receptor agonists and dipeptidyl peptidase-4 inhibitors in the treatment of diabetes: a review of clinical trials. Curr Opin Clin Nutr Metab Care 2008. 11:491-499. [DOD] [CrossRef]
- Hayes MR, Bradley L, Grill HJ. Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. Endocrinology 2009. 150:2654-2659. [DOD] [CrossRef]
- Ruttimann EB, Arnold M, Geary N, Langhans W. GLP-1 antagonism with exendin (9-39) fails to increase spontaneous meal size in rats. Physiol Behav 2010. 100:291-296. [DOD] [CrossRef]
- Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 2009. 150:1680-1687. [DOD] [CrossRef]
- Lehrke M, Marx N. Cardiovascular effects of incretin-based therapies. Rev Diabet Stud 2011. 8(3):382-391. [DOD] [CrossRef]
- Guzzardi MA, Iozzo P. Fatty heart, cardiac damage, and inflammation. Rev Diabet Stud 2011. 8(3):403-417. [DOD] [CrossRef]
- Holst JJ, Burcelin R, Nathanson E. Neuroprotective properties of GLP-1: theoretical and practical applications. Curr Med Res Opin 2011. 27:547-558. [DOD] [CrossRef]
- Perry T, Holloway HW, Weerasuriya A, Mouton PR, Duffy K, Mattison JA, Greig NH. Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp Neurol 2007. 203:293-301. [DOD] [CrossRef]
- Jin HY, Liu WJ, Park JH, Baek HS, Park TS. Effect of dipeptidyl peptidase-IV (DPP-IV) inhibitor (vildagliptin) on peripheral nerves in streptozotocin-induced diabetic rats. Arch Med Res 2009. 40:536-544. [DOD] [CrossRef]
- Liu WJ, Jin HY, Lee KA, Xie SH, Baek HS, Park TS. Neuroprotective effect of the glucagon-like peptide-1 receptor agonist, synthetic exendin-4, in streptozotocin-induced diabetic rats. Br J Pharmacol 2011. In press. [DOD]
- Jolivalt CG, Fineman M, Deacon CF, Carr RD, Calcutt NA. GLP-1 signals via ERK in peripheral nerve and prevents nerve dysfunction in diabetic mice. Diabetes Obes Metab 2011. In press. [DOD]
- Meier JJ, Gallwitz B, Siepmann N, Holst JJ, Deacon CF, Schmidt WE, Nauck MA. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003. 46:798-801. [DOD] [CrossRef]
- Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, Fujimoto S, Oku A, Tsuda K, Toyokuni S, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002. 8:738-742. [DOD] [CrossRef]
- Xie D, Zhong Q, Ding KH, Cheng H, Williams S, Correa D, Bollag WB, Bollag RJ, Insogna K, Troiano N, et al. Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone 2007. 40:1352-1360. [DOD] [CrossRef]
- Bollag RJ, Zhong Q, Phillips P, Min L, Zhong L, Cameron R, Mulloy AL, Rasmussen H, Qin F, Ding KH, Isales CM. Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology 2000. 141:1228-1235. [DOD] [CrossRef]
- Henriksen DB, Alexandersen P, Bjarnason NH, Vilsboll T, Hartmann B, Henriksen EE, Byrjalsen I, Krarup T, Holst JJ, Christiansen C. Role of gastrointestinal hormones in postprandial reduction of bone resorption. J Bone Miner Res 2003. 18:2180-2189. [DOD] [CrossRef]
- Rask E, Olsson T, Söderberg S, Johnson O, Seckl J, Holst JJ, Ahren B. Impaired incretin response after a mixed meal is associated with insulin resistance in nondiabetic men. Diabetes Care 2001. 24:1640-1645. [DOD] [CrossRef]
- Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, Holst JJ. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001. 86:3717-3723. [DOD] [CrossRef]
- Muscelli E, Mari A, Casolaro A, Camastra S, Seghieri G, Gastaldelli A, Holst JJ, Ferrannini E. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008. 57:1340-1348. [DOD] [CrossRef]
- Knop FK, Aaboe K, Vilsboll T, Madsbad S, Holst JJ, Krarup T. Reduced incretin effect in obese subjects with normal glucose tolerance as compared to lean control subjects. Diabetes 2008. 57(Suppl 1):A410. [DOD]
- Vollmer K, Holst JJ, Baller B, Ellrichmann M, Nauck MA, Schmidt WE, Meier JJ. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 2008. 57:678-687. [DOD] [CrossRef]
- Verdich C, Toubro S, Buemann B, Madsen JL, Holst JJ, Astrup A. The role of postprandial releases of insulin and incretin hormones in meal-induced satiety - effect of obesity and weight reduction. Int J Obes Relat Metab Disord 2001. 25:1206-1214. [DOD] [CrossRef]
- Ebert R, Creutzfeldt W. Gastric inhibitory polypeptide (GIP) hypersecretion in obesity depends on meal size and is not related to hyperinsulinemia. Acta Diabetol Lat 1992. 26:1-15. [DOD] [CrossRef]
- Amland PE, Jorde R, Burthol PG, Giercksky KE. Effect of intravenously infused porcine GIP on serum insulin in obese and lean subjects studied with the hyperglycemic clamp technique. Scand J Gastroenterol 1985. 20:309-314. [DOD] [CrossRef]
- Simonsson E, Ahren B. Potentiated beta-cell response to non-glucose stimuli in insulin-resistant C57BL/6J mice. Eur J Pharmacol 1998. 350:243-250. [DOD] [CrossRef]
- Nyholm B, Walker M, Gravholt CH, Shearing PA, Sturis J, Alberti KG, Holst JJ, Schmitz O. Twenty-four-hour insulin secretion rates, circulating concentrations of fuel substrates and gut incretin hormones in healthy offspring of Type II (non-insulin-dependent) diabetic parents: evidence of several aberrations. Diabetologia 1999. 42:1314-1323. [DOD] [CrossRef]
- Nauck MA, El-Ouaghlidi A, Gabrys B, Hücking K, Holst JJ, Deacon CF, Gallwitz B, Schmidt WE, Meier JJ. Secretion of incretin hormones (GIP and GLP-1) and incretin effect after oral glucose in first-degree relatives of patients with type 2 diabetes. Regul Pept 2004. 122:209-217. [DOD] [CrossRef]
- Vaag AA, Holst JJ, Volund A, Beck-Nielsen HB. Gut incretin hormones in identical twins discordant for non-insulin-dependent diabetes mellitus (NIDDM) - evidence for decreased glucagon-like peptide 1 secretion during oral glucose ingestion in NIDDM twins. Eur J Endocrinol 1996. 135:425-432. [DOD] [CrossRef]
- Bonde L, Vilsboll T, Nielsen T, Svare J, Holst JJ, Larsen S, Knop FK. Reduced postprandial GLP-1 response in gestational diabetes mellitus is a fully reversible phenomenon. Diabetes 2009. 58(Suppl 1):A372. [DOD]
- Meier JJ, Gallwitz B, Askenas M, Vollmer K, Deacon CF, Holst JJ, Schmidt WE, Nauck MA. Secretion of incretin hormones and the insulinotropic effect of gastric inhibitory polypeptide in women with a history of gestational diabetes. Diabetologia 2005. 48:1872-1881. [DOD] [CrossRef]
- Ahren B, Larsson H, Holst JJ. Reduced gastric inhibitory polypeptide but normal glucagon-like peptide 1 response to oral glucose in postmenopausal women with impaired glucose tolerance. Eur J Endocrinol 1997. 137:127-131. [DOD] [CrossRef]
- Muscelli E, Mari A, Natali A, Astiarraga BD, Camastra S, Frascerra S, Holst JJ, Ferrannini E. Impact of inretin hormones on beta-cell function in subjects with normal or impaired glucose tolerance. Am J Physiol Endocrinol Metab 2006. 291:1144-1150. [DOD] [CrossRef]
- Fritsche A, Stefan N, Hardt E, Häring H, Stumvoll M. Characterisation of beta-cell dysfunction of impaired glucose tolerance: evidence for impairment of incretin-induced insulin secretion. Diabetologia 2000. 43:852-858. [DOD] [CrossRef]
- Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986. 29:46-52. [DOD] [CrossRef]
- Bagger JI, Knop FK, Lund A, Vestergaard H, Holst JJ, Vilsboll T. Impaired regulation of the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab 2011. 96:737-345. [DOD] [CrossRef]
- Vilsboll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes 2001. 50:609-613. [DOD] [CrossRef]
- Ryskjaer J, Deacon CF, Carr RD, Krarup T, Madsbad S, Holst J, Vilsboll T. Plasma dipeptidyl peptidase-IV activity in patients with type-2 diabetes mellitus correlates positively with HbA1c levels, but is not acutely affected by food intake. Eur J Endocrinol 2006. 155:485-493. [DOD] [CrossRef]
- Deacon CF, Ahren B, Holst JJ. Inhibitors of dipeptidyl peptidase IV: A novel approach to prevention and treatment of type 2 diabetes? Expert Opin Investig Drugs 2004. 13:1091-1102. [DOD]
- Yasuda N, Inoue T, Nagakura T, Yamazaki K, Kira K, Saeki T, Tanaka I. Enhanced secretion of glucagon-like peptide 1 by biguanide compounds. Biochem Biophys Res Commun 2002. 298:779-784. [DOD] [CrossRef]
- Migoya EM, Bergeron R, Miller JL, Snyder RN, Tanen M, Hilliard D, Weiss B, Larson P, Gutierrez M, Jiang G, et al. Dipeptidyl peptidase-4 inhibitors administered in combination with metformin result in an additive increase in the plasma concentration of active GLP-1. Clin Pharmacol Ther 2010. 88:801-808. [DOD] [CrossRef]
- Yao X, Chen L, McIntyre MS, Danger DP, Nystrom CC, Smith CD, Young AA. Biguanide antidiabetic agents increase fecal bile acids via inhibition of apical sodium-dependent bile acid transporter. Diabetes 2010. 59(Suppl 1):A167. [DOD]
- Shang Q, Saumoy M, Holst JJ, Salen G, Xu G. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. Am J Physiol Gastrointest Liver Physiol 2010. 298:G419-G424. [DOD] [CrossRef]
- Garg SK, Ritchie PJ, Moser EG, Snell-Bergeon JK, Freson BJ, Hazenfield RM. Effects of colesevelam on LDL-C, A1c and GLP-1 levels in patients with type 1 diabetes: a pilot randomized double-blind trial. Diabetes Obes Metab 2011. 13:137-143. [DOD] [CrossRef]
- Ranganath L, Norris F, Morgan L, Wright J, Marks V. Delayed gastric emptying occurs following acarbose administration and is a further mechanism for its anti-hyperglycaemic effect. Diabet Med 1998. 15:120-124. [DOD] [CrossRef]
- Lee A, Patrick P, Wishart J, Horowitz M, Morley JE. The effects of miglitol on glucagon-like peptide-1 secretion and appetite sensations in obese type 2 diabetics. Diabetes Obes Metab 2002. 4:329-335. [DOD] [CrossRef]
- Moritoh Y, Takeuchi K, Hazama M. Chronic administration of voglibose, an alpha-glucosidase inhibitor, increases active glucagon-like peptide-1 levels by increasing its secretion and decreasing dipeptidyl peptidase-4 activity in ob/ob mice. J Pharmacol Exp Ther 2009. 329:669-676. [DOD] [CrossRef]
- Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia 2011. 54:10-18. [DOD]
- Kjems LL, Holst JJ, Vølund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 2003. 52:380-386. [DOD] [CrossRef]
- Hojberg PV, Vilsbøll T, Rabøl R, Knop FK, Bache M, Krarup T, Holst JJ, Madsbad S. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 2009. 52:199-207. [DOD] [CrossRef]
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 (7-36 amide) but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993. 91:301-307. [DOD] [CrossRef]
- Vilsboll T, Krarup T, Madsbad S, Holst JJ. Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia 2002. 45:1111-1119. [DOD] [CrossRef]
- Vilsboll T, Knop FK, Krarup T, Johansen A, Madsbad S, Larsen S, Hansen T, Pedersen O, Holst JJ. The pathophysiology of diabetes involves a defective amplification of the late-phase insulin response to glucose by glucose-dependent insulinotropic polypeptide-regardless of etiology and phenotype. J Clin Endocrinol Metab 2003. 88:4897-4903. [DOD] [CrossRef]
- Hansen KB, Vilsbøll T, Bagger JI, Holst JJ, Knop FK. Reduced glucose tolerance and insulin resistance induced by steroid treatment, relative physical inactivity, and high-calorie diet impairs the incretin effect in healthy subjects. J Clin Endocrinol Metab 2010. 95:3309-3317. [DOD] [CrossRef]
- Theodorakis MJ, Carlson O, Michopoulos S, Doyle ME, Juhaszova M, Petraki K, Egan JM. Human duodenal enteroendocrine cells: source of both incretin peptides, GLP-1 and GIP. Am J Physiol Endocrinol Metab 2006. 290:E550-E559. [DOD] [CrossRef]
- Knop FK, Vilsboll T, Højberg PV, Larsen S, Madsbad S, Vølund A, Holst JJ, Krarup T. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes 2007. 56:1951-1959. [DOD]
- Fukase N, Igarashi M, Takahashi H, Manaka H, Yamatani K, Daimon M, Tominaga M, Sasaki H. Hypersecretion of truncated glucagon-like peptide-1 and gastric inhibitory polypeptide in obese patients. Diabet Med 1993. 10:44-49. [DOD] [CrossRef]
- Salinari S, Bertuzzi A, Asnaghi S, Guidone C, Manco M, Mingrone G. First-phase insulin secretion restoration and differential response to glucose load depending on the route of administration in type 2 diabetic subjects after bariatric surgery. Diabetes Care 2009. 32:375-380. [DOD] [CrossRef]
This article has been cited by other articles:

|
Specificity and sensitivity of commercially available assays for glucagon-like peptide-1 (GLP-1): implications for GLP-1 measurements in clinical studies
Bak MJ, Wewer Albrechtsen NJ, Pedersen J, Knop FK, Vilsboll T, Jorgensen NB, Hartmann B, Deacon CF, Dragsted LO, Holst JJ
Diabetes Obes Metab 2014. 16(11):1155-1164
|
|

|
Gut hormones of preterm infants with abdominal symptoms and hypothyroxinemia
Kawamata R, Suzuki Y, Yada Y, Koike Y, Kono Y, Takahashi N
Pediatr Int 2014. In press
|
|

|
Accelerated intestinal glucose absorption in morbidly obese humans: relationship to glucose transporters, incretin hormones, and glycemia
Nguyen NQ, Debreceni TL, Bambrick JE, Chia B, Wishart J, Deane AM, Rayner CK, Horowitz M, Young RL
J Clin Endocrinol Metab 2014. In press
|
|
|
Additive effects of miglitol and anagliptin on insulin-treated type 2 diabetes mellitus: a case study
Kishimoto M, Noda M
Clin Drug Investig 2014. In press
|
|
|
Off-Label Antiobesity Treatment in Patients without Diabetes with GLP-1 Agonists in Clinical Practice
Sauer N, Reining F, Schulze Zur Wiesch C, Burkhardt T, Aberle J
Horm Metab Res 2014. In press
|
|

|
Rapid gastric and intestinal transit is a major determinant of changes in blood glucose, intestinal hormones, glucose absorption and postprandial symptoms after gastric bypass
Nguyen NQ, Debreceni TL, Bambrick JE, Bellon M, Wishart J, Standfield S, Rayner CK, Horowitz M
Obesity (Silver Spring) 2014. 22(9):2003-2009
|
|

|
Dietary sweet potato (Ipomoea batatas L.) leaf extract attenuates hyperglycaemia by enhancing the secretion of glucagon-like peptide-1 (GLP-1)
Nagamine R, Ueno S, Tsubata M, Yamaguchi K, Takagaki K, Hira T, Hara H, Tsuda T
Food Funct 2014. 5(9):2309-2316
|
|

|
Current challenges to overcome in the management of type 2 diabetes mellitus and associated neurological disorders
Khan NM, Ahmad A, Tiwari RK, Kamal MA, Mushtaq G, Ashraf GM
CNS Neurol Disord Drug Targets 2014. 13(8):1440-1457
|
|

|
Yellow pea fiber improves glycemia and reduces Clostridium leptum in diet-induced obese rats
Eslinger AJ, Eller LK, Reimer RA
Nutr Res 2014. 34(8):714-722
|
|
|
Diabetes drugs and neurological disorders: new views and therapeutic possibilities
Patrone C, Eriksson O, Lindholm D
Lancet Diabetes Endocrinol 2014. 2(3):256-262
|
|
|
Inhibition of phosphodiesterase 3B in insulin-secreting cells of normal and streptozocin-nicotinamide-induced diabetic rats: implications for insulin secretion
Zywert A, Szkudelska K, Szkudelski T
J Physiol Pharmacol 2014. 65(3):425-433
|
|
|
Effect of byetta on renal osmoregulatory function in patients with diabetes mellitus
Shakhmatova EI, Pimenova EV, Shchutskaia ZV
Eksp Klin Farmakol 2014. 77(3):24-27
|
|

|
Postprandial responses of incretin and pancreatic hormones in non-diabetic patients with end-stage renal disease
Idorn T, Knop FK, Jorgensen M, Holst JJ, Hornum M, Feldt-Rasmussen B
Nephrol Dial Transplant 2014. 29(1):119-127
|
|
|
Type 2 Diabetes Mellitus as a Neurocognitive Disorder: Pathophysiology and Therapeutic Approach
Kim J
J Korean Soc Biol Ther Psych 2014. 20(1):24-34
|
|

|
DPP-IV, An Important Target for Antidiabetic Functional Food Design
Ojeda MJ, Cereto-Massague A, Valls C, Pujadas G
Foodinformatics 2014. 2014:177-212
|
|

|
Measurement of the incretin hormones: glucagon-like peptide-1 and glucose-dependent insulinotropic peptide
Kuhre RE, Wewer Albrechtsen NJ, Hartmann B, Deacon CF, Holst JJ
J Diabetes Complications 2014. pii:S1056-8727(14)00402-4
|
|

|
A novel GIP-oxyntomodulin hybrid peptide acting through GIP, glucagon and GLP-1 receptors exhibits weight reducing and anti-diabetic properties
Bhat VK, Kerr BD, Flatt PR, Gault VA
Biochem Pharmacol 2013. 85(11):1655-1662
|
|

|
A new endoscopically implantable device (SatiSphere) for treatment of obesity - efficacy, safety, and metabolic effects on glucose, insulin, and GLP-1 levels
Sauer N, Rösch T, Pezold J, Reining F, Anders M, Groth S, Schachschal G, Mann O, Aberle J
Obes Surg 2013. 23(11):1727-1733
|
|

|
Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta-analysis of clinical studies
Calanna S, Christensen M, Holst JJ, Laferrere B, Gluud LL, Vilsboll T, Knop FK
Diabetes Care 2013. 36(10):3346-3352
|
|

|
Glucose Metabolism in Critically Ill Patients: Are Incretins an Important Player?
Nielsen ST, Krogh-Madsen R, Moller K
J Intensive Care Med 2013. In press
|
|

|
Pleiotropic effects of incretins and antidiabetic drugs with incretine mechanism
Winkler G
Orv Hetil 2013. 154(7):248-255
|
|

|
Glucose-lowering effect of the DPP-4 inhibitor sitagliptin after glucose and non-glucose macronutrient ingestion in non-diabetic subjects
Ohlsson L, Alsalim W, Carr RD, Tura A, Pacini G, Mari A, Ahrén B
Diabetes Obes Metab 2013. 15(6):531-537
|
|

|
Enteroendocrine hormone mimetics for the treatment of obesity and diabetes
Irwin N, Flatt PR
Curr Opin Pharmacol 2013. 13(6):989-995
|
|

|
Drugs or diet? - Developing novel therapeutic strategies targeting the free fatty acid family of GPCRs
Dranse HJ, Kelly ME, Hudson BD
Br J Pharmacol 2013. 170(4):696-711
|
|

|
Incretin hormones and beta-cell mass expansion: what we know and what is missing?
Tortosa F, Dotta F
Arch Physiol Biochem 2013. 119(4):161-169
|
|

|
Incretin dysfunction in type 2 diabetes: clinical impact and future perspectives
Ahren B
Diabetes Metab 2013. 39(3):195-201
|
|

|
Val(8) GLP-1-Glu-PAL: a GLP-1 agonist that improves hippocampal neurogenesis, glucose homeostasis, and beta-cell function in high-fat-fed mice
Lennox R, Porter DW, Flatt PR, Gault VA
ChemMedChem 2013. 8(4):595-602
|
|
|
Peripheral Pathways in the Food-Intake Control towards the Adipose-Intestinal Missing Link
Mendieta Zeron H, Dominguez Garcia MV, Camarillo Romero Mdel S, Flores-Merino MV
Int J Endocrinol 2013. 2013:598203
|
|
|
Follow-up observation of therapeutic effects of gastric bypass on type 2 diabetes patients with overweight,obesity and non-obesity in China
Gao HK, Jia YL, Wu ZW, et al.
Chin J Diabetes 2012. 2012:4
|
|

|
Inhibition of Dipeptidyl Peptidase-4 (DPP-4): A Target to Treat Type 2 Diabetes
Ahren B
Curr Enzyme Inhibit 2011. 7(4):205-217
|
|

|
The alpha-cell as target for type 2 diabetes therapy
Christensen M, Bagger JI, Vilsboll T, Knop F
Rev Diabet Stud 2011. 8(3):369-381
|
|

|
The continuing need for drug development and clinical trials in type 2 diabetes and its complications: introduction to the RDS Special Issue
Raz I, Gallwitz B
Rev Diabet Stud 2011. 8(3):288-292
|
|
|


)
)
)
)

