Review
| Rev Diabet Stud,
2012,
9(1):6-22 |
DOI 10.1900/RDS.2012.9.6 |
Genetic and environmental factors associated with type 2 diabetes and diabetic vascular complications
Mariana Murea, Lijun Ma, Barry I. Freedman
Department of Internal Medicine, Section on Nephrology, Wake Forest School of Medicine, Winston-Salem, North Carolina, USA
Address correspondence to: Mariana Murea, email: mmurea@wakehealth.edu
Manuscript submitted February 13, 2012; resubmitted April 24, 2012; accepted April 27, 2012.
Keywords: type 2 diabetes, gene, environment, vascular complications, polymorphism, heritability, GWAS, linkage analysis
Abstract
Faced with a global epidemic of type 2 diabetes (T2D), it is critical that researchers improve our understanding of the pathogenesis of T2D and related vascular complications. These findings may ultimately lead to novel treatment options for disease prevention or delaying progression. Two major paradigms jointly underlie the development of T2D and related coronary artery disease, diabetic nephropathy, and diabetic retinopathy. These paradigms include the genetic risk variants and behavioral/environmental factors. This article systematically reviews the literature supporting genetic determinants in the pathogenesis of T2D and diabetic vasculopathy, and the functional implications of these gene variants on the regulation of beta-cell function and glucose homeostasis. We update the discovery of diabetes and diabetic vasculopathy risk variants, and describe the genetic technologies that have uncovered them. Also, genomic linkage between obesity and T2D is discussed. There is a complementary role for behavioral and environmental factors modulating the genetic susceptibility and diabetes risk. Epidemiological and clinical data demonstrating the effects of behavioral and novel environmental exposures on disease expression are reviewed. Finally, a succinct overview of recent landmark clinical trials addressing glycemic control and its impact on rates of vascular complications is presented. It is expected that novel strategies to exploit the gene- and exposure-related underpinnings of T2D will soon result.
Abbreviations: AA - African American; ACACB - acetyl-coenzyme A carboxylase beta; ADIPOQ - adiponectin gene; AGA - advanced glycation end-product; ATM - ataxia-telangiectasia mutation; ATZ - atrazine; BMI - body mass index; CNDP1(2) - carnosinase 1(2); CNV - common copy number variation; CAD - coronary artery disease; CARS - cysteinyl-tRNA synthetase; CKD - chronic kidney disease; DN - diabetic nephropathy; DPP - Diabetes Prevention Program; DR - diabetic retinopathy; DZ - dizygotic; EA - European American; ELMO1 - engulfment and cell motility 1; eNOS - endothelial nitric oxide synthase; EPO - erythropoietin; ESRD - end-stage renal disease; EWAS - environment-wide association study; FIND - Family Investigation of Nephropathy and Diabetes; FERM - moesin; FRMD3 - moesin domain containing 3; GLO1 - glyoxalase 1; GLP-1 - glucagon-like peptide-1; GSTM1(T1) - glutathione S-transferases M1 (T1); GSV - Genome Structural Variation (Consortium); GWAS - genome-wide association study; HIV - human immunodeficiency virus; HOMA-IR - homeostasis model assessment of insulin resistance; HR - hazard ratio; KCNJ11 - potassium inwardly rectifying channel subfamily J member 11; LEPR - leptin receptor; LIMK2 - LIM domain kinase 2; MAF - minor allele frequency; MODY - maturity-onset diabetes of the young; mRNA - messenger ribonucleic acid; MTNR1B - melatonin receptor type 1 B; MZ - monozygotic; NF-κB - nuclear factor kappa-light-chain-enhancer of activated B cells; NO2 - nitrogen dioxide; OR - odds ratio; PCB - polychlorinated biphenyl; PKCB1 - PKC beta 1; PKC-zeta - protein kinase C isoform zeta; PM - particulate matter; PPAR - peroxisome proliferator-activated receptor; PRKCB1 - protein kinase C beta 1; PVT1 - plasmacytoma variant translocation 1; RAGE - receptor for advanced glycation end products; RPS12 - ribosomal protein S12; SFI1 - spindle assembly associated; SNP - single nucleotide polymorphism; SOD1 - superoxide dismutase 1; T2D - type 2 diabetes; TCF7L2 - transcription factor 7-like 2; TNFAIP3 - tumor necrosis factor induced protein 3
Morbidity and mortality associated with type 2 diabetes
The prevalence of type 2 diabetes (T2D) has been steadily rising in recent decades. Worldwide, the number of diagnosed diabetes cases reached 30 million in 1985, 135 million in 1995, 366 million in 2011, and is predicted to reach 552 million by 2030. The prevalence of T2D vastly exceeds that of type 1 diabetes, accounting for up to 90% of all diabetes cases (http://diabetes.niddk.nih.gov/dm/pubs/statistics/index.aspx#fast, accessed February, 2012).
Diabetes is the seventh leading cause of death in the United States. Compared with the general non-diabetic population, persons with diabetes have approximately a 7-year shorter life expectancy, an effect directly related to the major diabetic complications [1]. Patients with diabetes develop macrovascular complications (including coronary artery disease (CAD), peripheral vascular disease, and stroke) and microvascular complications (including diabetic nephropathy (DN), diabetic retinopathy (DR), and peripheral neuropathy), which are collectively known as diabetic vascular complications. Worldwide, diabetes is the major cause of CAD, limb amputations, end-stage renal disease (ESRD), and blindness. Compared to non-diabetic individuals, adults with diabetes have a 2-4-fold higher death rate from CAD, and those with ESRD on dialysis have a 4-fold reduction in life expectancy [2, 3]. A recent study investigated the association of diabetes with a broad range of health conditions. People with diabetes had a 25-75% higher risk of dying from cancer, infections, liver disease, lung disease, mental disorders, intentional self-harm, external causes, and falls, independent of other risk factors (such as age, gender, smoking, and weight). Overall, approximately 40% of the excess deaths in diabetic patients appears to be due to non-vascular events [4].
Classification of type 2 diabetes
Recent advances in molecular and genetic research have enabled a classification of T2D into clinical sub-phenotypes of various pathophysiology. A small fraction of all diabetes cases (1-5%), termed maturity-onset diabetes of the young (MODY), occur as a result of single-gene mutations impacting the ability of the pancreatic β-cells to produce or secrete insulin [5]. Diabetes can also develop secondary to a specific disease or as a result of exogenous factors such as:
- medications (e.g., corticosteroids, calcineurin inhibitors, thiazide diuretics, antipsychotics, HIV protease inhibitors, statins),
- viral infections (e.g., hepatitis C), and
- hormonal factors, including pregnancy and oral contraceptives [6].
These types can be classified as "other specific types of diabetes". Notably, only a subset of individuals exposed to exogenous triggers develop diabetes.
Increased individual risk of diabetes may be explained by one or more of the following factors:
- Variable loads of adverse environmental exposures.
- Congenitally reduced pancreatic beta-cell mass.
- Host genetic susceptibility.
The most prevalent form of diabetes, T2D per se, accounts for more than 80% of diagnosed cases. It represents a heterogenous entity which does not include type 1 diabetes, genetic forms of MODY, gestational diabetes, or other specific types secondary to specific diseases or exposures [6]. It is generally agreed that T2D is a multifactorial disease, arising from the presence of T2D risk alleles in multiple genes and a disease-conducive environment. Environmental factors modulating genetic risk are proving to be important in the development of T2D.
Heritability and environment in type 2 diabetes
Several lines of evidence support the principle of inherited genetic susceptibility as an important risk factor for common T2D. The offspring of a parent with T2D face a 40% life-time risk of developing T2D, increasing to 70% when both parents have T2D [7]. The mode of genetic transmission of common T2D, unlike monogenic diabetes, does not follow simple Mendelian patterns. Twin studies have assessed the relative importance of heredity and environment on the etiology of T2D. In a population-based cohort of twins in Finland, the cumulative concordance rate of T2D was clearly higher among monozygotic (MZ) twins (34% probandwise, 20% pairwise) than dizygotic (DZ) twins (16% probandwise, 8% pairwise), while the cumulative incidence of diabetes did not differ between MZ and DZ twins [8]. Importantly, this study applied threshold modeling techniques to estimate and compare the contribution of heritability, shared environmental, and unique environmental sources of variation in the underlying liability to T2D. Based on best fit models, heritability was 46%, shared environmental effects were 15%, and unshared environmental effects accounted for 38% of the liability for T2D [8].
The risk of T2D is higher in certain ethnic groups, independent of metabolic risk factor profiles. For example, Pima Indians have a 10-fold higher prevalence of T2D than the general U.S. population. Based on 2004-2006 national survey data for individuals 20 years of age or older, 6.6% of non-Hispanic whites, 7.5% of Asian Americans, 10.4% of Hispanic Americans, and 11.8% of non-Hispanic blacks had diagnosed T2D. (http://diabetes.niddk.nih.gov/dm/pubs/statistics/#Racial, accessed February, 2012). However, shared or unique environmental and genetic effects may be possible reasons for ethnic differences in susceptibility to T2D.
Finding the genetic determinants of common type 2 diabetes - methods, results, and functions
Genetic research tools have evolved significantly in recent decades. Table 1 contains the principal genetic methods and the presumed mechanism of action of the major genes reproducibly associated with T2D and related glycemic traits.
Table
1.
Major genes associated with type 2 diabetes and related traits, based on study methodology and putative functional effects |
|
 |
 |
Candidate gene association studies and linkage analysis were the primary methods used to identify T2D susceptibility loci, prior to the application of genome-wide association studies (GWAS). In the candidate gene approach, genes were localized in pathways that were considered likely to be involved in the pathogenesis of diabetes, including PPAR-gamma (PPARG) and potassium inwardly rectifying channel subfamily J member 11 (KCNJ11). The protein products of these two genes are targeted by antidiabetic medications (thiazolidinediones and sulphonylureas) and were found to harbor missense mutations associated with T2D risk [9, 10].
Family-based linkage analyses have helped to identify causal mutations underpinning the Mendelian forms of diabetes in MODY [11]. Among families with common form of T2D, genome-wide linkage scans have identified several T2D linkage peaks on chromosomes 1q21, 10q25.3, and 20q13.3. The most replicated region across different cohorts and ethnic groups resides on chromosome 1q21-25 [12-17]. Further analysis of the chromosome 10 region showed that variants in the transcription factor 7-like 2 (TCF7L2) gene were strongly associated with T2D in multiple independent cohorts [18]. TCF7L2 is a component of the Wnt signaling pathway, and it is a transcriptional regulator involved in stimulating the proliferation of pancreatic β-cells, regulating embryonic development of the pancreatic mass, and controlling the production of the incretin hormone glucagon-like peptide-1 (GLP-1) in intestinal endocrine cells [19].
Major advancements in unveiling genetic determinants of T2D were made through GWAS, a methodology using single nucleotide polymorphism (SNP) markers across the genome. The outcomes of GWAS have helped in identifying novel pathways, and highlighted the role of β-cell dysfunction in T2D. The prevailing theory of the pathobiology of common T2D implicates an interplay of defective insulin secretion, defective insulin response (insulin resistance), and environmental factors (e.g., westernized food pattern, obesity, and sedentary life style). Experimentally, excess adipose tissue and increased plasma free fatty acid levels disrupt insulin signal transduction and glucose uptake in insulin-sensitive tissues, a phenomenon termed lipotoxicity [20]. Obesity and diets rich in saturated fatty acids can induce T2D through β-cell endoplasmic reticulum stress and apoptosis [21]. Adipose tissue is also a source of circulating free fatty acids, cytokines, and other proteins that are mechanistically related to insulin resistance [22]. Although it was anticipated that T2D genetic markers would point toward genes with impacts on insulin action, most T2D risk alleles identified through GWAS have impacts on glucose-induced insulin secretion (ADAMTS9, ADCY5, CDC123/CAMK1D, CDKAL1, CDKN2A/B, CENTD2, DGKB/TMEM195, FOXO1, GCK, HHEX, IGF2BP2, KCNQ1, KCNJ11, MTNR1B, PROX1, SGK1, SLC30A8, TCF7L2, TSPAN8/LGR5, THADA). Only a few genes have effects on insulin resistance (FTO, GCKR, KLF14, PPARG, ADAMTS 9), and the functions of many associated variants remain unknown (Table 1) [23].
There has been much speculation about potential mechanisms by which gene polymorphisms affect insulin secretion. KCJN11 and SGK1 encode or activate β-cell potassium channels, respectively. Polymorphisms in these genes may increase the probability of the channel opening or closing, and thereby impact the cellular mechanism of insulin secretion [24]. Experimentally, genetic variants of KCNQ1, TCF7L2, GIPR, and WFS1 can affect glucose-induced incretin hormone release in the intestine and incretin-stimulated insulin release in the pancreas [25].
It is also important to note that the glycemic phenotype can depend on the nature of genetic variation within and across different ethnic groups. Germline loss-of-function mutations in the PPARG gene have been detected in familial forms of diabetes where cases exhibit severe insulin resistance and hypertension [26]. In contrast, the widespread polymorphism (Pro12Ala allele) is protective against T2D in Caucasians, but not in South Asians [9, 27]. Similarly, different mutations in HNF4A or IPF1 can lead to either pancreatic agenesis (for homozygous dominant-negative frameshift mutation in IPF1), MODY (for heterozygous dominant-negative frameshift mutation in IPF1, or loss of function mutation in HNF4A), or late-onset diabetes (for missense mutations in IPF1 or HNF4A) [28-30]. Studies examining prediabetic glycemic traits (fasting insulin, fasting glucose, glucose, and insulin levels after glucose challenge) have identified that loci implicated in T2D susceptibility can also harbor variants that correlate with continuous glycemic traits. However, loci associated with elevated fasting blood glucose levels have not uniformly been associated with T2D, suggesting that elevations in fasting glucose do not necessarily herald future risk of T2D, and that the pathophysiologic mechanisms of pre-diabetes (elevated fasting blood glucose) may differ from those necessary to induce overt T2D [31]. In conclusion, severe mutations can cause marked β-cell dysfunction and monogenic diabetes with autosomal dominant Mendelian inheritance within the same gene; whereas, common polymorphisms can modulate the risk of impaired glucose tolerance and T2D.
Type 2 diabetes and obesity - genetic ties
Since the rising prevalence of common T2D paralleled that of obesity, it was assumed that the genomic signals found in T2D studies could broadly overlap with the genomic signals captured in obesity studies. However, only three replicated loci have been found to be shared in both obesity and T2D (FTO, PREX1, and MTNR1B) (Figure 1). The GWAS associations between FTO and PREX1 polymorphism and T2D were driven by obesity, since statistical adjustment for body mass index (BMI) eliminated the associations with diabetes [32, 33]. On the other hand, common variants in melatonin receptor type 1 B gene (MTNR1B) are associated with the risk of T2D independent of BMI [34, 35]. Receptors for melatonin (MTNR1A and MTNR1B) are expressed in pancreatic islet cells, and carriers of the risk genotype exhibit impaired glucose-stimulated insulin secretion [36, 37].
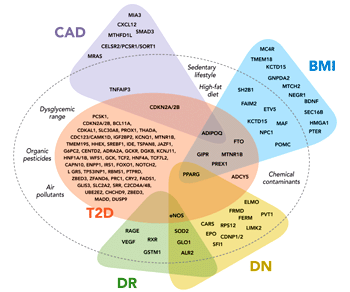 |
|
Figure 1. Interplay between genes and environmental/behavioral factors in the development of type 2 diabetes and related vascular complications. Variability in diabetes and obesity-related genes predispose to type 2 diabetes (T2D) and diabetic vascular complications such as coronary artery disease (CAD), diabetic nephropathy (DN), and diabetic retinopathy (DR). Behavioral (e.g., sedentary lifestyle, high-fat diet) and environmental factors (e.g., organic pesticides, chemical exposures, and air pollutants) have complementary effects on the development of type 2 diabetes. Additional genes are associated with CAD and body mass index (BMI) in the general population, without demonstrable effects on the risk of T2D. |
|
As aforementioned, the Pro12Ala polymorphism in PPARG can modulate the risk of T2D, and functional differences between proline- and alanine-containing gene products have been identified. PPARG is a transcription factor with a pivotal role in adipogenesis. PPARG expression is increased in adipose tissue of obese subjects [38]. Individuals with Pro12Ala substitution in PPARG have lower BMI and improved insulin sensitivity. In vitro studies revealed that the protective Pro isoform has higher transcriptional activity [39]. These data suggest that the Pro12Ala variant may be a genetic factor that contribute to both obesity and insulin resistance.
The "missing heritability" in common type 2 diabetes
To date, GWAS have identified nearly 52 common risk variants that associate with T2D [40, 41]. However, most of the risk alleles detected by GWAS have weak impacts and are located in intronic (non-coding) segments of genes or within chromosomal regions that do not contain genes (gene deserts). Together, these genes capture only 10% of the estimated heritability in T2D. Barring the association of T2D with the transcription factor 7-like 2 gene (TCF7L2), with odds ratio (OR) of 1.4 (each risk allele increases risk of T2D by approximately 40%), the remaining T2D risk variants generally have odds ratios (ORs) well below 1.3. Moreover, statistical prediction models using nearly 20 susceptibility loci did not confer superior prediction of disease, compared to traditional metabolic risk factors [42-44]. An even more comprehensive analysis, based on 40 autosomal SNPs, captured only an additional 10% of T2D-prone individuals aged less than 50 years, compared with a prediction based only on clinical factors. These 40 SNPs did not provide superior predictive ability in individuals over age 50 [45]. A general statistical model based on risk variants, each with an OR of 1.3-1.5 per allele, suggested that 50 or more independent risk alleles would be necessary to predict disease development with nearly 90% accuracy [46].
In view of these complexities, the concept of "missing heritability" emerged along with novel hypotheses for the presence of other genetic determinants (common copy number variations (CNVs), rare variants), or the interplay of different factors (epistasis, gene-environment interaction), to account for the unexplained heritability [47]. For example, a DNA copy number variation at the leptin receptor (LEPR) gene locus is associated with metabolic syndrome-related traits and risk of T2D. Lower LEPR copy numbers related to higher fasting glucose; and LEPR expression in lymphoblastoid cell lines correlated with the number of CNVs [48]. However, a role for common CNVs in the risk of T2D remains unclear. A genome-wide CNV-typing array of 3,432 common polymorphic CNVs, covering nearly 90% of total CNVs with minor allele frequencies (MAF) >5%, was designed and applied to nearly 3,000 T2D cases and 2,000 non-diabetic controls in the Genome Structural Variation Consortium (GSV). The results revealed that a CNV locus in TSPAN8 was associated with T2D, which was previously identified by SNP GWAS. This suggests that T2D gene associations are more frequently driven by SNPs than CNVs [49].
By design, GWAS captures common allelic variants across the genome, occurring in more than 5% of the population. A shift in the focus has evolved towards the possible contribution of low frequency (>1% and <5% of the population) or rare frequency (<1% of the population) variants to account for the unexplained heritability. The prime technology for detecting rare variants is direct sequencing, which may:
1. target regions of interest defined by GWAS with strong and repeated replication,
2. sequence all protein-coding regions ('exomes'), or
3. sequence the entire genome [47].
It has also been proposed that gene-gene interactions between T2D susceptibility variants could be nonlinear (in terms of epistasis), and thus jointly increase the genetic effect. However, despite broad acceptance of this theory, it has been difficult to detect interactions between SNPs. A recent study exploring main and epistatic effects in 70,236 markers tagging common SNPs showed that, although there was a two-locus association at 79 SNP-pairs encompassing TCF7L2 SNP paired with variants in FTO, TSPAN8, and CDKAL1 (Bonferroni-corrected p-value = 0.05), the actual joint two-locus effects could be attributed to single-locus association at TCF7L2 [50]. Nevertheless, it is assumed that a very strong single-locus effect could conceal multilocus findings. Therefore, it is necessary that methodological approaches undergo further developments to explore the epistatic phenomena.
Behavioral and environmental factors
Sedentary lifestyles and high-fat diets are behavioral factors conducive to the high prevalence of T2D. Two large randomized cohorts of lifestyle interventions (dietary modification, weight loss, and exercise) in overweight adults (mean BMI 31 kg/m2) with impaired glucose tolerance achieved 58% reduction in the incidence of diabetes. These results were directly associated with lifestyle changes and weight loss [51, 52]. Nevertheless, approximately 10% of overweight adults with impaired glucose tolerance progressed to T2D within 3 years in these studies, despite rigorous lifestyle modification. While these cases of T2D could be due to the presence of non-modifiable genetic risk factors, it led to the theory that earlier lifestyle intervention may be necessary. In the future, long-term results from lifestyle intervention programs applied in school-aged individuals high at risk of T2D could shed more light on a possible time-dependent environment effect [53].
A recently discussed environmental risk factor for T2D is pollution. Epidemiological data indicate that chronic exposure to organic land pollutants (pesticides, herbicides) disturbs glucose metabolism and induces insulin resistance. A strong dose-dependent relationship between serum concentrations of organic pesticides (subclasses of organochlorine and non-dioxin-like polychlorinated biphenyls) and prevalence of diabetes has been demonstrated, with ORs up to 11.5 for prevalent diabetes among those with the highest percentiles of serum pesticide levels compared to those with undetectable levels [54]. Serum levels of organochlorine pesticides in the general (non-diabetic) population are associated with humoral markers of insulin resistance (homeostasis model assessment of insulin resistance (HOMA-IR) derived from fasting glucose and insulin levels). This association is stronger in overweight individuals with the highest serum pesticide concentration and absent in those with the lowest concentration [55]. Because of the lipophilic nature of the organic pollutants, obesity may serve as a storage compartment for these toxic chemicals which induce insulin resistance and T2D in a dose- and possibly time-dependent manner [56, 57]. These findings have biologic plausibility exemplified in several experimental models. Exposure to the herbicide atrazine (ATZ) can cause mitochondrial toxicity and promote obesity and insulin resistance, with even more detrimental effects observed when animals were fed a high-fat diet [58]. Pesticides can suppress the expression of functional glucose transporter proteins in several organs, providing a hypothetical mechanism for the observed link between these chemicals and insulin resistance [59].
Air pollutants have also been studied in relation to the prevalence of T2D. Exposure to traffic-related pollutants (particulate matter (PM) and nitrogen dioxide (NO2)) is associated with higher incidence rates of T2D in a dose dependent manner. The risk is present predominantly in women. The highest exposures carry a >20% increase in T2D prevalence after adjusting for other risk covariates (e.g., obesity, population density, ethnicity, income, education, smoking, health insurance, and indicators of indoor pollutant exposures) [60-63]. Plausible mechanisms by which air pollutants contribute to the development of T2D include induction of inflammation in adipose tissue and insulin resistance, demonstrated to occur in animals exposed to PM [64].
Other chemical contaminants in food (dioxins and polychlorinated biphenyls (PCBs)) or water (arsenic), and occupational exposures to various toxins (N-nitroso compounds, benzene, polycyclic aromatic hydrocarbons, and solvents in the rubber industry, e.g. dioxins and talc among pulp and paper mill workers, tetrachloroethylene among dry-cleaning workers, and N-nitroso compounds in engine manufacturing workers) have been linked with excess risk of T2D and mortality from diabetes. However, these data remain inconclusive because of the limited number of studies, small samples, and possible publication biases (reviewed in reference [65]).
As oxidative stress plays a role in insulin resistance, it was hypothesized that antioxidants might confer protection against T2D [66]. An inverse association between serum carotenoids, plasma vitamin C, and plasma tocopherol concentrations (markers of fruit and vegetable intake, or dietary supplements) with T2D and impaired glucose metabolism has been detected [67-69]. Also, a direct relation between vitamin E and vitamin C intake and insulin sensitivity has been reported among healthy supplement users [70, 71].
A recent study employed a novel methodology akin to GWAS, whereby environmental factors purported to cause T2D (pesticides, heavy metals, consumption of dietary supplements) were modeled using a method termed "environment-wide association study" (EWAS). Significant risk associations were identified between higher systemic levels of the pesticide derivative heptachlor epoxide, polychlorinated biphenyl, and gamma-tocopherol (vitamin E), and lower beta-carotene (vitamin A precursor) levels [72].
Gene-environment interactions
Evidence that environmental factors modulate phenotypic expression emerged from the Diabetes Prevention Program (DPP) [52]. This multi-ethnic cohort was designed to test the preventive effects of a lifestyle intervention (low-fat diet, exercise, and behavior modification) or medication (metformin) on progression to diabetes in high-risk individuals with impaired glucose tolerance. Remarkably, for every 6.9 or 13.9 persons enrolled in the lifestyle-intervention or metformin program, respectively, one case of T2D was prevented. This translates into a significant 58% and 31% reduction, respectively, in the incidence of T2D. In a post-hoc analysis genotyping nearly 3,500 participants for transcription factor 7-like 2 (TCF7L2) polymorphisms (rs12255372 and rs7903146), the effect of the risk-conferring genotype on progression to diabetes was at its peak in the placebo group (hazard ratio (HR) 1.81, p = 0.004), less in the metformin group (HR 1.62, p = 0.04), and the risk dissipated in the lifestyle-intervention group (HR 1.15, p = 0.60). The results were similar among the homozygous carriers of either of the two risk alleles, indicating that a behavioral intervention can mitigate the risk conferred by the genetic background [73]. A more recent analysis included genotyping of up to 34 T2D risk loci in DPP participants to examine whether the preventive interventions maintained their effectiveness in those individuals with higher genetic risk burden based on the computation of a genetic risk score. Importantly, treatment with either metformin or lifestyle intervention was effective in reducing the risk of diabetes at any level of genetic risk, with a trend towards the possibility that lifestyle intervention is more effective in individuals with the highest genetic risk scores [74].
Gene-environment interactions can be defined as genetic effects on (disease) traits that differ in magnitude (and sometimes direction) across environmental contexts. In most GWAS studies, it is assumed that the genetic effects are equal across the spectrum of the environmental exposure (i.e., no interactions exist). If this assumption is incorrect, then clinically relevant genetic risk factors may be missed [75]. This is the case for ataxia-telangiectasia mutated (ATM) variants that were associated with glycemic response to metformin in T2D. Whereas, no existing GWAS has revealed a significant association between ATM variants and diabetes, inhibition of ATM attenuates the phosphorylation and activation of AMP-activated protein kinase in response to metformin [76].
Heritability and environmental factors in diabetic vascular complications
Genetic and environmental factors play important roles in the development of T2D and diabetic vascular complications (Figure 1).
Coronary artery disease
Family history of CAD adds to cardiovascular risk in patients with diabetes, implicating either shared environmental factors and/or genetic factors [77, 78]. Marked racial differences are seen in the prevalence of subclinical atherosclerosis, manifested by levels of coronary artery, thoracic aorta, and abdominal aorta calcified atherosclerotic plaque. Relative to European Americans (EAs), the prevalence and severity of coronary calcification are markedly lower in African Americans (AAs) (52% vs. 70%). The same applies to the prevalence and severity of aortic and thoracic calcification (63% vs. 80%, and 27% vs. 42%), when adjusted for multiple CV risk factors [79, 80]. Carotid artery intima-media thickness in families with T2D has an estimated heritability approximating 40%, and ethnicity (AA vs. EA) is an important predictor [81].
To date, the best validated genetic risk marker for CAD in the general population is the chromosome 9p21.3 locus. Individuals homozygous for the CAD risk allele on chromosome 9p21 have a 40% increased risk of CAD and an estimated risk of suffering myocardial infarction 1.64 times greater than non-carriers [82]. In a case-control study of nearly 1,200 T2D patients from two different cohorts, the 9p21 variant was associated with CAD risk, and the risk was increased by history of poor glycemic control [83]. Diabetic subjects with two 9p21 risk alleles have a doubled odds for CAD at adequate glycemic control, and quadrupled odds for CAD at poor glycemic control. Of interest, previous studies have shown that different SNPs at the chromosome 9p21.3 locus contribute to the risk of T2D (Figure 1) [84]. Since large-vessel atherosclerosis can precede the development of diabetes (unlike classical microvascular complications), it had long been postulated that both conditions may share common genetic and environmental traits (dyslipidemia, hypertension), rather than atherosclerosis being a complication of diabetes [85]. A recent large-scale case-control study in a Chinese population was the first to report that two SNPs (rs10811661-T and rs10757283-C) on the 9p21.3 locus contribute to both the risk of T2D (OR 1.23 and 1.30, respectively) and CAD (OR 1.19-1.18, respectively) [86].
Adiponectin is a hormone with anti-atherogenic and anti-inflammatory effects, and variants in the adiponectin gene (ADIPOQ) have been assessed for association with CAD in diabetics. Homozygotes of +276T allele have a 30-80% lower risk of CAD [87, 88], while homozygotes of allele -4034C have an approximately 60% increased risk of CAD [88]. However, other studies showed conflicting results, with either opposing effects of the +45G allele on CAD risk [87, 89], or the presence of early-onset CAD in association with SNP +276T [90]. No association has been found between variants +276T and +45G with the development of T2D, although allele -11377G may be a risk factor for T2D (OR 1.12, 95%CI 1.02-1.23, p = 0.009, dominant model) [91].
At the molecular level, atherosclerosis is characterized by chronic inflammation and activation of transcription factor nuclear factor-κB (NF-κB) [92]. Tumor necrosis factor induced protein 3 (TNFAIP3) is a cytoplasmic protein that limits inflammation by inhibition of NF-κB [93]. Polymorphisms in the TNFAIP3 gene were shown to be independently associated with CAD, with an adjusted OR of 2.3, and paradoxical higher OR of 3.9 in individuals with good glycemic control. Peripheral blood mononuclear cells carrying risk alleles had 30-45% lower mRNA TNFAIP3 levels [94]. In an attempt to explain this paradoxical finding, it was argued that cellular pathways mediating the increased risk of CAD in T2D may differ in the two glycemic states. While NF-κB-mediated mechanisms may be predominant in the presence of moderate hyperglycemia, it is speculated that other pathways may prevail in the presence of more severe hyperglycemia [94].
Diabetic nephropathy
It is generally accepted that there is a genetic susceptibility to DN. Albuminuria, chronic kidney disease (CKD), and ESRD segregate within diabetic families, but DN develops only in a subset of T2D-affected populations in all race groups [95, 96]. Also, there is a racial affinity with DN, which further underscores the presence of genetic and/or environmental risk factors. African Americans (AAs) with T2D have an approximately 40% higher risk of DN and 3 times higher rates of progression to ESRD, relative to European Americans (EAs) [97, 98]. Several genes have been reproducibly associated with DN susceptibility (Figure 1):
- erythropoietin (EPO)
- superoxide dismutase 1 (SOD1),
- 4.1 protein,
- ezrin,
- radixin,
- moesin (FERM) domain containing 3 (FRMD3),
- cysteinyl-tRNA synthetase (CARS),
- plasmacytoma variant translocation 1 (PVT1),
- engulfment and cell motility 1 (ELMO1),
- carnosinase 1 (CNDP1),
- carnosinase 2 (CNDP2),
- endothelial nitric oxide synthase (eNOS),
- acetyl-coenzyme A carboxylase beta (ACACB),
- protein kinase C beta 1 (PRKCB1),
- LIM domain kinase 2 (LIMK2),
- Sfi1 homolog,
- spindle assembly associated (SFI1), and
- ribosomal protein S12 (RPS12) [99-102].
The in vivo disease mechanisms driven by these polymorphisms are intensively studied. Similar to genetic pathways in the development of CAD, it can be theorized that some T2D mechanistic pathways may share a common pathogenesis with susceptibility to DN. For example, a single nucleotide polymorphism in the ACACB gene is strongly associated with albuminuria and advanced CKD in Asians and EAs [101, 103]. The gene product is the acetyl-CoA carboxylase beta enzyme, which is important in fatty acid oxidation. The latter is a cellular process implicated in insulin resistance via the mechanism of lipotoxicity [104]. Also, the protein kinase C isoform zeta (PKC-zeta) gene modulates inflammatory pathways in adipose cells. PKC-zeta deficiency generates insulin resistance in vivo [105]. Polymorphisms in the PKC beta 1 (PKCB1) gene are associated with increased risk of ESRD in Chinese subjects with T2D [100]. The Pro12Ala polymorphism in the human peroxisome proliferator-activated receptor gamma (PPARG) gene is associated with improved insulin sensitivity, decreased risk of T2D, and lower levels of albuminuria and DN [106, 107].
Diabetic retinopathy
Although the risk of diabetic retinopathy (DR) increases with diabetes duration and poor glycemic control, some patients develop DR in spite of adequate glycemic control. Familial clustering of DR cases across different ethnicities suggests that genetic susceptibility may underlie this risk [108, 109]. The genetic heritability of DR in the FIND-Eye cohort was estimated at 25% [110]. Retinopathy develops in approximately 21% of patients newly diagnosed with T2D and in approximately 60% of patients with T2D within the first 2 decades after diagnosis [111].
There is a strong association between microalbuminuria and DR in persons with T2D. The prevalence of DR is doubled in patients with microalbuminuira, and 6 times higher in the presence of macroalbuminuria [112], suggesting that DN and DR may share common risk factors, including genetic susceptibility factors. Indeed, several genes have been associated with DR and DN [113-115].
The pathophysiology of DR is complex, and involves the interplay of numerous molecular mediators, such as advanced glycation end products (AGEs), oxidative stress, neoangiogenesis, and activation of the polyol pathway. Accumulation of AGEs in diabetes affects the extracellular matrix and a variety of cellular functions implicated in diabetic vascular complications by engaging the receptor for advanced glycation end products (RAGE) [116]. Polymorphisms in the RAGE gene are associated with an increased risk of DR, particularly in East Asians [117, 118].
Oxidative stress plays an important role in the development of DR [119]. In this regard, it was investigated whether polymorphisms in the glutathione S-transferases M1 (GSTM1) and T1 (GSTT1) genes, encoding enzymes with a role in repairing endogenous compounds affected by oxidative stress, are associated with DR. A case-control study including 604 Caucasians with T2D found that deletion of GSTT1 was twice as common in T2D with DR than in diabetics without DR, while deletion of the GSTM1 was half as common in patients with DR [120]. Polymorphisms of the gene encoding the glyoxalase 1 (GLO1) enzyme that dispose endogenous toxins can also confer susceptibility to DR and DN [113]. Superoxide dismutase 2 (SOD2) is an enzyme with critical defense against oxidative stress. Several studies in different ethnic groups found that a polymorphism in the SOD2 gene confers protection from DR and DN [115]. Endothelial nitric oxide synthase (eNOS) and ischemia-induced retinal neovascularization also play a role in the pathogenesis of proliferative DR [121], and presence of the eNOS 4a/4a homozygous deletion was associated with a 3.4 times (95% CI 1.4-8.6, p = 0.009) increased risk of PDR when adjusted for other risk factors [122].
Angiogenic factors such as vascular endothelial growth factor (VEGF) play a pivotal role in the pathogenesis of DR. Common polymorphisms in the promoter region, 5'-untranslated region (UTR) and 3'UTR of the VEGF gene were strongly associated with elevated risk of DR [123]. Anti-angiogenic VEGF inhibitors, including bevacizumab and pegaptanib, have been used in the treatment of proliferative DR in small trials. In the short term, the proportion of anti-VEGF-treated patients with complete regression of retinal neovascularization was significantly higher than in the control groups [124, 125]. Polymorphism in the retinoid-X receptor (RXR) gamma gene has also been associated with DR (OR = 2.388; 95% confidence interval = 1.17-4.875) in Asians with T2D [126]. Polymorphisms in the regulatory region of the aldose reductase (ALR2) gene (encoding the rate-limiting enzyme of the polyol pathway) were associated with susceptibility to DN and DR [114, 127]. However, the impacts of these genetic risk factors were modest, and some studies yielded inconsistent findings [128, 129].
The role of hyperglycemic environmental exposure in vascular complications
The strong relationship between hyperglycemia and diabetic vascular complications is the legacy of diabetes and its medical management. Hyperglycemia induces endothelial dysfunction through direct toxic effects (hyperosmolarity and advanced glycation end products), and through derangements in multiple interrelated intracellular signaling pathways (e.g., aldose reductase pathway, advanced glycation end product pathway, reactive oxygen intermediate pathway, and protein kinase C pathway) [130]. In summary, these alterations result in an amplification of endothelial proliferative factors (insulin-like growth factor 1, transforming growth factor β, and vascular endothelial growth factor), activation of the NF-κB pathway, and vascular inflammation; all of which underlie vascular diseases in diabetes.
Although glycemic control early in the course of diabetes reduces vascular complication rates, end-organ vascular complications still occur at high frequencies. This observation led to the theory of persistent epigenetic changes that contribute to diabetic vascular complications long after the occurrence of the hyperglycemic events because of hyperglycemia-associated histone modifications. These modifications persist after restoration of normoglycemia, a process termed 'hyperglycemic memory' or 'legacy effect' [131]. Histone modifications (altered acetylation, methylation, phosphorylation, and/or sumoylation) control transcription expression, cell cycle regulation, and cellular differentiation [132, 133]. In vitro studies in endothelial cell lines demonstrated that cells exposed to hyperglycemia underwent histone modification, activation of multiple transcription factors [134], increased H3K4 methylation at the NF-κB-p65 promoter, enhanced expression of NF-κB-p65, and increased NF-κB-driven pro-inflammatory gene expression [135].
Importantly, the duration of hyperglycemia can differentially impact the susceptibility to diabetes-related microvascular and macrovascular complications. This conclusion is based on results from several large randomized intensive vs. conventional therapy trials for blood sugar control in patients with T2D. The design and results of four landmark trials (UKPDS, ADVANCE, ACCORD, and VADT) are summarized in Table 2. In UKPDS, ADVANCE, and ACCORD, protection from microvascular disease (DR and/or albuminuria as surrogate of DN) was achieved with tight glycemic control [136-139]. In VADT, no clear benefit of glycemic control on microvascular complications was detected. However, this study had fewer patients with long diabetes durations prior to enrollment and a shorter follow-up period [140].
Table
2.
Baseline characteristics and vascular outcomes stratified by tight (T) and conventional (C) glycemic control |
|
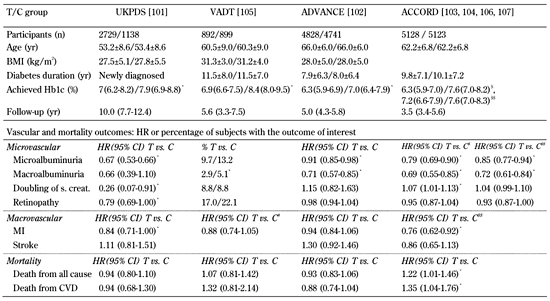 |
|
Legend:
Data are number, mean ± SD, median (interquartile range), percentage, or HR (95% CI) in tight control (T) group vs. cnventional control (C) group. * p < 0.05. # Composite CV events. $ Outcomes at 3.5 years. $$ Outcomes at 5 years, after transition from T to C glycemic control in the intensive-therapy group. Abbreviations: UKPDS - UK Prospective Diabetes Study, VADT - Veterans Affairs Diabetes Trial, ADVANCE - Action in Diabetes and Vascular Disease, ACCORD - Action to Control Cardiovascular Risk in Diabetes, BMI - body mass index, HbA1c - glycated hemoglobin, MI - myocardial infarction, HR - hazard ratio, CI - confidence interval, SD - standard deviation, s. creat. - serum creatinine. |
|
The relationship between good glycemic control and protection from macrovascular complications has been critically questioned. In ADVANCE and VADT, significant declines in the incidence rates of cardiovascular disease through improved glucose control could not be detected [137, 140]. The only study showing declines in cardiovascular events through tight glucose control was UKPDS [136]. More surprising were the ACCORD results, with a higher mortality observed in the intensive control group [141]. The negative impact of tight glycemic control was maintained in the long term follow-up, further suggesting the presence of a glycemic memory phenomenon in diabetic vascular complications [142].
The different macrovascular complication rates may be caused by important design differences between ACCORD, ADVANCE, and UKPDS (Table 2). ACCORD and ADVANCE enrolled patients with longstanding diabetes (mean diabetes duration 8-12 years) and shorter follow-up durations (mean follow-up 5 years), as opposed to newly diagnosed subjects with T2D and 10 year follow-up in UKPDS. Also, baseline burdens of cardiovascular disease risk factors, medication regimens for diabetes control, rates of hypoglycemia, blood pressure and lipid control, and definitions used for the primary vascular outcomes were heterogenous across these studies, and could underlie the variability of results. Despite the problems associated with the different study designs, the results provided support for the ‘legacy effect’ in the case of macrovascular complications. Persistent hyperglycemia induces deleterious consequences in the vasculature which perpetuate even in the event of subsequent improvements in glucose control. These results underscore the need for early detection and appropriate therapeutic intervention in diabetes, with the need for moderate glycemic control (HbA1c 6.2-7%), as opposed to tight control (HbA1c ≤ 6.1%).
Concluding remarks
The clinical and pathophysiologic spectrum of T2D has expanded greatly with the detection of genetic determinants through high-throughput genetic studies. Although a susceptible genetic background appears to be necessary for the development of overt diabetes, it is only fully sufficient in rare Mendelian forms of diabetes. Western lifestyles, obesity, dietary supplements, exposure to organic pollutants, and glycemic control serve as examples of non-genetic environmental factors which ultimately interact with risk alleles in susceptibility genes to initiate the common forms of T2D and diabetic vascular complications. As opposed to the presently immutable and complex genetic heritability, exogenous risk factors are amendable by intervention, which may save a considerable proportion of patients from development of T2D and vascular complications.
GWAS results are used as a platform for gene-environment and gene-gene interaction studies. In the future, new avenues will be developed to reveal the intricate genetic architecture of complex diseases by performing targeted genome sequencing, whole-exome sequencing, or whole genome sequencing. The current data reflect decades of functional work to decipher the underlying biology. It is hoped that this research will ultimately translate into new therapeutic modalities.
Disclosure: The authors report no conflict of interests in relation to this work.
References
- Morgan CL, Currie CJ, Peters JR. Relationship between diabetes and mortality: a population study using record linkage. Diabetes Care 2000. 23:1103-1107. [DOD] [CrossRef]
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998. 339:229-234. [DOD] [CrossRef]
- Goodkin DA, Bragg-Gresham JL, Koenig KG, Wolfe RA, Akiba T, Andreucci VE, Saito A, Rayner HC, Kurokawa K, Port FK, et al. Association of comorbid conditions and mortality in hemodialysis patients in Europe, Japan, and the United States: the Dialysis Outcomes and Practice Patterns Study (DOPPS). J Am Soc Nephrol 2003. 14:3270-3277. [DOD] [CrossRef]
- Seshasai SR, Kaptoge S, Thompson A, Di AE, Gao P, Sarwar N, Whincup PH, Mukamal KJ, Gillum RF, Holme I, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med 2011. 364:829-841. [DOD] [CrossRef]
- Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med 2010. 16:407-416. [DOD] [CrossRef]
- Diagnosis and classification of diabetes mellitus. Diabetes Care 2010. 33(Suppl 1):S62-S69. [DOD]
- Meigs JB, Cupples LA, Wilson PW. Parental transmission of type 2 diabetes: the Framingham Offspring Study. Diabetes 2000. 49:2201-2207. [DOD] [CrossRef]
- Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, Stengard J, Kesaniemi YA. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 1992. 35:1060-1067. [DOD] [CrossRef]
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 2000. 26:76-80. [DOD] [CrossRef]
- Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003. 52:568-572. [DOD] [CrossRef]
- Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 2001. 345:971-980. [DOD] [CrossRef]
- Vionnet N, Hani EH, Dupont S, Gallina S, Francke S, Dotte S, De MF, Durand E, Lepretre F, Lecoeur C, et al. Genomewide search for type 2 diabetes-susceptibility genes in French whites: evidence for a novel susceptibility locus for early-onset diabetes on chromosome 3q27-qter and independent replication of a type 2-diabetes locus on chromosome 1q21-q24. Am J Hum Genet 2000. 67:1470-1480. [DOD] [CrossRef]
- Wiltshire S, Hattersley AT, Hitman GA, Walker M, Levy JC, Sampson M, O'Rahilly S, Frayling TM, Bell JI, Lathrop GM, et al. A genomewide scan for loci predisposing to type 2 diabetes in a U.K. population (the Diabetes UK Warren 2 Repository): analysis of 573 pedigrees provides independent replication of a susceptibility locus on chromosome 1q. Am J Hum Genet 2001. 69:553-569. [DOD] [CrossRef]
- Meigs JB, Panhuysen CI, Myers RH, Wilson PW, Cupples LA. A genome-wide scan for loci linked to plasma levels of glucose and HbA(1c) in a community-based sample of Caucasian pedigrees: The Framingham Offspring Study. Diabetes 2002. 51:833-840. [DOD] [CrossRef]
- Hsueh WC, St Jean PL, Mitchell BD, Pollin TI, Knowler WC, Ehm MG, Bell CJ, Sakul H, Wagner MJ, Burns DK, et al. Genome-wide and fine-mapping linkage studies of type 2 diabetes and glucose traits in the Old Order Amish: evidence for a new diabetes locus on chromosome 14q11 and confirmation of a locus on chromosome 1q21-q24. Diabetes 2003. 52(2):550-557. [DOD] [CrossRef]
- Xiang K, Wang Y, Zheng T, Jia W, Li J, Chen L, Shen K, Wu S, Lin X, Zhang G, et al. Genome-wide search for type 2 diabetes/impaired glucose homeostasis susceptibility genes in the Chinese: significant linkage to chromosome 6q21-q23 and chromosome 1q21-q24. Diabetes 2004. 53:228-234. [DOD] [CrossRef]
- Ng MC, So WY, Lam VK, Cockram CS, Bell GI, Cox NJ, Chan JC. Genome-wide scan for metabolic syndrome and related quantitative traits in Hong Kong Chinese and confirmation of a susceptibility locus on chromosome 1q21-q25. Diabetes 2004. 53:2676-2683. [DOD] [CrossRef]
- Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 2006. 38:320-323. [DOD] [CrossRef]
- Jin T, Liu L. The Wnt signaling pathway effector TCF7L2 and type 2 diabetes mellitus. Mol Endocrinol 2008. 22:2383-2392. [DOD] [CrossRef]
- Cusi K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr Diab Rep 2010. 10:306-315. [DOD] [CrossRef]
- Morgan NG. Fatty acids and beta-cell toxicity. Curr Opin Clin Nutr Metab Care 2009. 12:117-122. [DOD] [CrossRef]
- Lazar MA. How obesity causes diabetes: not a tall tale. Science 2005. 307:373-375. [DOD] [CrossRef]
- Imamura M, Maeda S. Genetics of type 2 diabetes: the GWAS era and future perspectives. Endocr J 2011. 58:723-739. [DOD] [CrossRef]
- Schafer SA, Machicao F, Fritsche A, Haring HU, Kantartzis K. New type 2 diabetes risk genes provide new insights in insulin secretion mechanisms. Diabetes Res Clin Pract 2011. 93(Suppl 1):S9-S24. [DOD] [CrossRef]
- Mussig K, Staiger H, Machicao F, Haring HU, Fritsche A. Genetic variants affecting incretin sensitivity and incretin secretion. Diabetologia 2010. 53:2289-2297. [DOD] [CrossRef]
- Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999. 402:880-883. [DOD]
- Radha V, Vimaleswaran KS, Babu HN, Abate N, Chandalia M, Satija P, Grundy SM, Ghosh S, Majumder PP, Deepa R, et al. Role of genetic polymorphism peroxisome proliferator-activated receptor-gamma2 Pro12Ala on ethnic susceptibility to diabetes in South-Asian and Caucasian subjects: Evidence for heterogeneity. Diabetes Care 2006. 29:1046-1051. [DOD] [CrossRef]
- Hani EH, Suaud L, Boutin P, Chevre JC, Durand E, Philippi A, Demenais F, Vionnet N, Furuta H, Velho G, et al. A missense mutation in hepatocyte nuclear factor-4 alpha, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. J Clin Invest 1998. 101:521-526. [DOD] [CrossRef]
- Macfarlane WM, Frayling TM, Ellard S, Evans JC, Allen LI, Bulman MP, Ayres S, Shepherd M, Clark P, Millward A, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J Clin Invest 1999. 104:R33-R39. [DOD] [CrossRef]
- Hani EH, Stoffers DA, Chevre JC, Durand E, Stanojevic V, Dina C, Habener JF, Froguel P. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J Clin Invest 1999. 104:R41-R48. [DOD] [CrossRef]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010. 42:105-116. [DOD] [CrossRef]
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007. 316:889-894. [DOD] [CrossRef]
- Lewis JP, Palmer ND, Ellington JB, Divers J, Ng MC, Lu L, Langefeld CD, Freedman BI, Bowden DW. Analysis of candidate genes on chromosome 20q12-13.1 reveals evidence for BMI mediated association of PREX1 with type 2 diabetes in European Americans. Genomics 2010. 96:211-219. [DOD] [CrossRef]
- Andersson EA, Holst B, Sparso T, Grarup N, Banasik K, Holmkvist J, Jorgensen T, Borch-Johnsen K, Egerod KL, Lauritzen T, et al. MTNR1B G24E variant associates With BMI and fasting plasma glucose in the general population in studies of 22,142 Europeans. Diabetes 2010. 59:1539-1548. [DOD] [CrossRef]
- Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M, Delplanque J, Lobbens S, Rocheleau G, Durand E, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 2009. 41:89-94. [DOD] [CrossRef]
- Ramracheya RD, Muller DS, Squires PE, Brereton H, Sugden D, Huang GC, Amiel SA, Jones PM, Persaud SJ. Function and expression of melatonin receptors on human pancreatic islets. J Pineal Res 2008. 44:273-279. [DOD] [CrossRef]
- Staiger H, Machicao F, Schafer SA, Kirchhoff K, Kantartzis K, Guthoff M, Silbernagel G, Stefan N, Haring HU, Fritsche A. Polymorphisms within the novel type 2 diabetes risk locus MTNR1B determine beta-cell function. PLoS One 2008. 3:e3962. [DOD] [CrossRef]
- Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, Flier JS. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest 1997. 99:2416-2422. [DOD] [CrossRef]
- Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet 1998. 20:284-287. [DOD] [CrossRef]
- Wheeler E, Barroso I. Genome-wide association studies and type 2 diabetes. Brief Funct Genomics 2011. 10:52-60. [DOD] [CrossRef]
- Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 2012. 44:67-72. [DOD] [CrossRef]
- Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D'Agostino RB Sr. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med 2007. 167:1068-1074. [DOD] [CrossRef]
- Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 2008. 359:2220-2232. [DOD] [CrossRef]
- Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, Manning AK, Florez JC, Wilson PW, D'Agostino RB Sr, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med 2008. 359:2208-2219. [DOD] [CrossRef]
- de Miguel-Yanes JM, Shrader P, Pencina MJ, Fox CS, Manning AK, Grant RW, Dupuis J, Florez JC, D'Agostino RB Sr, Cupples LA, et al. Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care 2011. 34:121-125. [DOD] [CrossRef]
- Janssens AC, Aulchenko YS, Elefante S, Borsboom GJ, Steyerberg EW, van Duijn CM. Predictive testing for complex diseases using multiple genes: fact or fiction? Genet Med 2006. 8:395-400. [DOD]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 2010. 11:446-450. [DOD] [CrossRef]
- Jeon JP, Shim SM, Nam HY, Ryu GM, Hong EJ, Kim HL, Han BG. Copy number variation at leptin receptor gene locus associated with metabolic traits and the risk of type 2 diabetes mellitus. BMC Genomics 2010. 11:426. [DOD] [CrossRef]
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 2010. 464:713-720. [DOD] [CrossRef]
- Bell JT, Timpson NJ, Rayner NW, Zeggini E, Frayling TM, Hattersley AT, Morris AP, McCarthy MI. Genome-wide association scan allowing for epistasis in type 2 diabetes. Ann Hum Genet 2011. 75:10-19. [DOD] [CrossRef]
- Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001. 344:1343-1350. [DOD] [CrossRef]
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002. 346:393-403. [DOD] [CrossRef]
- Foster GD, Linder B, Baranowski T, Cooper DM, Goldberg L, Harrell JS, Kaufman F, Marcus MD, Trevino RP, Hirst K. A school-based intervention for diabetes risk reduction. N Engl J Med 2010. 363:443-453. [DOD] [CrossRef]
- Lee DH, Lee IK, Song K, Steffes M, Toscano W, Baker BA, Jacobs DR Jr. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999-2002. Diabetes Care 2006. 29:1638-1644. [DOD] [CrossRef]
- Lee DH, Lee IK, Jin SH, Steffes M, Jacobs DR Jr. Association between serum concentrations of persistent organic pollutants and insulin resistance among nondiabetic adults: results from the National Health and Nutrition Examination Survey 1999-2002. Diabetes Care 2007. 30:622-628. [DOD] [CrossRef]
- Porta M. Persistent organic pollutants and the burden of diabetes. Lancet 2006. 368:558-559. [DOD] [CrossRef]
- Lee DH, Jacobs DR Jr, Porta M. Could low-level background exposure to persistent organic pollutants contribute to the social burden of type 2 diabetes? J Epidemiol Community Health 2006. 60:1006-1008. [DOD]
- Lim S, Ahn SY, Song IC, Chung MH, Jang HC, Park KS, Lee KU, Pak YK, Lee HK. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS One 2009. 4:e5186. [DOD] [CrossRef]
- Olsen H, Enan E, Matsumura F. Regulation of glucose transport in the NIH 3T3 L1 preadipocyte cell line by TCDD. Environ Health Perspect 1994. 102:454-458. [DOD] [CrossRef]
- Brook RD, Jerrett M, Brook JR, Bard RL, Finkelstein MM. The relationship between diabetes mellitus and traffic-related air pollution. J Occup Environ Med 2008. 50:32-38. [DOD] [CrossRef]
- Pearson JF, Bachireddy C, Shyamprasad S, Goldfine AB, Brownstein JS. Association between fine particulate matter and diabetes prevalence in the U.S. Diabetes Care 2010. 33:2196-2201. [DOD] [CrossRef]
- Kramer U, Herder C, Sugiri D, Strassburger K, Schikowski T, Ranft U, Rathmann W. Traffic-related air pollution and incident type 2 diabetes: results from the SALIA cohort study. Environ Health Perspect 2010. 118:1273-1279. [DOD] [CrossRef]
- Coogan PF, White LF, Jerrett M, Brook RD, Su JG, Seto E, Burnett R, Palmer JR, Rosenberg L. Air pollution and incidence of hypertension and diabetes mellitus in black women living in los angeles. Circulation 2012. 125:767-772. [DOD] [CrossRef]
- Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, Mikolaj MB, Cai Y, Ostrowski MC, Lu B, Parthasarathy S, et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 2009. 119:538-546. [DOD] [CrossRef]
- Longnecker MP, Daniels JL. Environmental contaminants as etiologic factors for diabetes. Environ Health Perspect 2001. 109(Suppl 6):871-876. [DOD]
- Ceriello A. Oxidative stress and glycemic regulation. Metabolism 2000. 49:27-29. [DOD] [CrossRef]
- Coyne T, Ibiebele TI, Baade PD, Dobson A, McClintock C, Dunn S, Leonard D, Shaw J. Diabetes mellitus and serum carotenoids: findings of a population-based study in Queensland, Australia. Am J Clin Nutr 2005. 82:685-693. [DOD]
- Salonen JT, Nyyssonen K, Tuomainen TP, Maenpaa PH, Korpela H, Kaplan GA, Lynch J, Helmrich SP, Salonen R. Increased risk of non-insulin dependent diabetes mellitus at low plasma vitamin E concentrations: a four year follow up study in men. BMJ 1995. 311:1124-1127. [DOD] [CrossRef]
- Sargeant LA, Wareham NJ, Bingham S, Day NE, Luben RN, Oakes S, Welch A, Khaw KT. Vitamin C and hyperglycemia in the European Prospective Investigation into Cancer - Norfolk (EPIC-Norfolk) study: a population-based study. Diabetes Care 2000. 23:726-732. [DOD] [CrossRef]
- Facchini F, Coulston AM, Reaven GM. Relation between dietary vitamin intake and resistance to insulin-mediated glucose disposal in healthy volunteers. Am J Clin Nutr 1996. 63:946-949. [DOD]
- Feskens EJ, Virtanen SM, Rasanen L, Tuomilehto J, Stengard J, Pekkanen J, Nissinen A, Kromhout D. Dietary factors determining diabetes and impaired glucose tolerance. A 20-year follow-up of the Finnish and Dutch cohorts of the Seven Countries Study. Diabetes Care 1995. 18:1104-1112. [DOD] [CrossRef]
- Patel CJ, Bhattacharya J, Butte AJ. An Environment-Wide Association Study (EWAS) on type 2 diabetes mellitus. PLoS One 2010. 5:e10746. [DOD] [CrossRef]
- Florez JC, Jablonski KA, Bayley N, Pollin TI, de Bakker PI, Shuldiner AR, Knowler WC, Nathan DM, Altshuler D. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med 2006. 355(3):241-250. [DOD] [CrossRef]
- Hivert MF, Jablonski KA, Perreault L, Saxena R, McAteer JB, Franks PW, Hamman RF, Kahn SE, Haffner S, Meigs JB, et al. Updated genetic score based on 34 confirmed type 2 diabetes Loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes 2011. 60:1340-1348. [DOD] [CrossRef]
- Franks PW. Gene x environment interactions in type 2 diabetes. Curr Diab Rep 2011. 11:552-561. [DOD] [CrossRef]
- Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, Donnelly LA, Schofield C, Groves CJ, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet 2011. 43:117-120. [DOD] [CrossRef]
- Wagenknecht LE, Bowden DW, Carr JJ, Langefeld CD, Freedman BI, Rich SS. Familial aggregation of coronary artery calcium in families with type 2 diabetes. Diabetes 2001. 50(4):861-866. [DOD] [CrossRef]
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010. 3:97-105. [DOD] [CrossRef]
- Bild DE, Detrano R, Peterson D, Guerci A, Liu K, Shahar E, Ouyang P, Jackson S, Saad MF. Ethnic differences in coronary calcification: the Multi-Ethnic Study of Atherosclerosis (MESA). Circulation 2005. 111(10):1313-1320. [DOD] [CrossRef]
- Allison MA, Budoff MJ, Nasir K, Wong ND, Detrano R, Kronmal R, Takasu J, Criqui MH. Ethnic-specific risks for atherosclerotic calcification of the thoracic and abdominal aorta (from the Multi-Ethnic Study of Atherosclerosis). Am J Cardiol 2009. 104:812-817. [DOD] [CrossRef]
- Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke 2002. 33(7):1876-1881. [DOD] [CrossRef]
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007. 316:1491-1493. [DOD] [CrossRef]
- Doria A, Wojcik J, Xu R, Gervino EV, Hauser TH, Johnstone MT, Nolan D, Hu FB, Warram JH. Interaction between poor glycemic control and 9p21 locus on risk of coronary artery disease in type 2 diabetes. JAMA 2008. 300:2389-2397. [DOD] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007. 447:661-678. [DOD] [CrossRef]
- Stern MP. Diabetes and cardiovascular disease. The "common soil" hypothesis. Diabetes 1995. 44(4):369-374. [DOD] [CrossRef]
- Cheng X, Shi L, Nie S, Wang F, Li X, Xu C, Wang P, Yang B, Li Q, Pan Z, et al. The same chromosome 9p21.3 locus is associated with type 2 diabetes and coronary artery disease in a Chinese Han population. Diabetes 2011. 60:680-684. [DOD] [CrossRef]
- Bacci S, Menzaghi C, Ercolino T, Ma X, Rauseo A, Salvemini L, Vigna C, Fanelli R, Di MU, Doria A, et al. The +276 G/T single nucleotide polymorphism of the adiponectin gene is associated with coronary artery disease in type 2 diabetic patients. Diabetes Care 2004. 27:2015-2020. [DOD] [CrossRef]
- Qi L, Doria A, Manson JE, Meigs JB, Hunter D, Mantzoros CS, Hu FB. Adiponectin genetic variability, plasma adiponectin, and cardiovascular risk in patients with type 2 diabetes. Diabetes 2006. 55:1512-1516. [DOD] [CrossRef]
- Lacquemant C, Froguel P, Lobbens S, Izzo P, Dina C, Ruiz J. The adiponectin gene SNP+45 is associated with coronary artery disease in Type 2 (non-insulin-dependent) diabetes mellitus. Diabet Med 2004. 21:776-781. [DOD] [CrossRef]
- Filippi E, Sentinelli F, Romeo S, Arca M, Berni A, Tiberti C, Verrienti A, Fanelli M, Fallarino M, Sorropago G, et al. The adiponectin gene SNP+276G>T associates with early-onset coronary artery disease and with lower levels of adiponectin in younger coronary artery disease patients (age [DOD] [CrossRef]
- Han LY, Wu QH, Jiao ML, Hao YH, Liang LB, Gao LJ, Legge DG, Quan H, Zhao MM, Ning N, et al. Associations between single-nucleotide polymorphisms (+45T>G, +276G>T, -11377C>G, -11391G>A) of adiponectin gene and type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia 2011. 54:2303-2314. [DOD] [CrossRef]
- Rask-Madsen C, King GL. Proatherosclerotic mechanisms involving protein kinase C in diabetes and insulin resistance. Arterioscler Thromb Vasc Biol 2005. 25:487-496. [DOD] [CrossRef]
- Heyninck K, Beyaert R. A20 inhibits NF-kappaB activation by dual ubiquitin-editing functions. Trends Biochem Sci 2005. 30:1-4. [DOD] [CrossRef]
- Boonyasrisawat W, Eberle D, Bacci S, Zhang YY, Nolan D, Gervino EV, Johnstone MT, Trischitta V, Shoelson SE, Doria A. Tag polymorphisms at the A20 (TNFAIP3) locus are associated with lower gene expression and increased risk of coronary artery disease in type 2 diabetes. Diabetes 2007. 56:499-505. [DOD] [CrossRef]
- Maeda S. Genetics of diabetic nephropathy. Ther Adv Cardiovasc Dis 2008. 2:363-371. [DOD] [CrossRef]
- Freedman BI, Bostrom M, Daeihagh P, Bowden DW. Genetic factors in diabetic nephropathy. Clin J Am Soc Nephrol 2007. 2:1306-1316. [DOD] [CrossRef]
- Crook ED, Patel SR. Diabetic nephropathy in African-American patients. Curr Diab Rep 2004. 4:455-461. [DOD] [CrossRef]
- Crook ED, Wofford P, Oliver B. Advanced diabetic nephropathy disproportionately affects African-American females: cross-sectional analysis and determinants of renal survival in an academic renal clinic. Ethn Dis 2003. 13:28-33. [DOD]
- Divers J, Freedman BI. Susceptibility genes in common complex kidney disease. Curr Opin Nephrol Hypertens 2010. 19:79-84. [DOD] [CrossRef]
- Ma RC, Tam CH, Wang Y, Luk AO, Hu C, Yang X, Lam V, Chan AW, Ho JS, Chow CC, et al. Genetic variants of the protein kinase C-beta 1 gene and development of end-stage renal disease in patients with type 2 diabetes. JAMA 2010. 304:881-889. [DOD] [CrossRef]
- Maeda S, Kobayashi MA, Araki S, Babazono T, Freedman BI, Bostrom MA, Cooke JN, Toyoda M, Umezono T, Tarnow L, et al. A single nucleotide polymorphism within the acetyl-coenzyme A carboxylase beta gene is associated with proteinuria in patients with type 2 diabetes. PLoS Genet 2010. 6:e1000842. [DOD] [CrossRef]
- Freedman BI, Langefeld CD, Lu L, Divers J, Comeau ME, Kopp JB, Winkler CA, Nelson GW, Johnson RC, Palmer ND, et al. Differential effects of MYH9 and APOL1 risk variants on FRMD3 association with diabetic ESRD in African Americans. PLoS Genet 2011. 7:e1002150. [DOD] [CrossRef]
- Tang SC, Leung VT, Chan LY, Wong SS, Chu DW, Leung JC, Ho YW, Lai KN, Ma L, Elbein SC, et al. The acetyl-coenzyme A carboxylase beta (ACACB) gene is associated with nephropathy in Chinese patients with type 2 diabetes. Nephrol Dial Transplant 2010. 25(12):3931-3934. [DOD] [CrossRef]
- Murea M, Freedman BI, Parks JS, Antinozzi PA, Elbein SC, Ma L. Lipotoxicity in diabetic nephropathy: the potential role of Fatty Acid oxidation. Clin J Am Soc Nephrol 2010. 5:2373-2379. [DOD] [CrossRef]
- Lee SJ, Kim JY, Nogueiras R, Linares JF, Perez-Tilve D, Jung DY, Ko HJ, Hofmann SM, Drew A, Leitges M, et al. PKCzeta-regulated inflammation in the nonhematopoietic compartment is critical for obesity-induced glucose intolerance. Cell Metab 2010. 12:65-77. [DOD] [CrossRef]
- Liu L, Zheng T, Wang F, Wang N, Song Y, Li M, Li L, Jiang J, Zhao W. Pro12Ala polymorphism in the PPARG gene contributes to the development of diabetic nephropathy in Chinese type 2 diabetic patients. Diabetes Care 2010. 33:144-149. [DOD] [CrossRef]
- Caramori ML, Canani LH, Costa LA, Gross JL. The human peroxisome proliferator-activated receptor gamma2 (PPARgamma2) Pro12Ala polymorphism is associated with decreased risk of diabetic nephropathy in patients with type 2 diabetes. Diabetes 2003. 52:3010-3013. [DOD] [CrossRef]
- Rema M, Saravanan G, Deepa R, Mohan V. Familial clustering of diabetic retinopathy in South Indian type 2 diabetic patients. Diabet Med 2002. 19:910-916. [DOD] [CrossRef]
- Hallman DM, Huber JC Jr, Gonzalez VH, Klein BE, Klein R, Hanis CL. Familial aggregation of severity of diabetic retinopathy in Mexican Americans from Starr County, Texas. Diabetes Care 2005. 28:1163-1168. [DOD] [CrossRef]
- Arar NH, Freedman BI, Adler SG, Iyengar SK, Chew EY, Davis MD, Satko SG, Bowden DW, Duggirala R, Elston RC, et al. Heritability of the severity of diabetic retinopathy: the FIND-Eye study. Invest Ophthalmol Vis Sci 2008. 49:3839-3845. [DOD] [CrossRef]
- Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL 3rd, Klein R. Diabetic retinopathy. Diabetes Care 2003. 26:226-229. [DOD] [CrossRef]
- Rani PK, Raman R, Gupta A, Pal SS, Kulothungan V, Sharma T. Albuminuria and Diabetic Retinopathy in Type 2 Diabetes Mellitus Sankara Nethralaya Diabetic Retinopathy Epidemiology And Molecular Genetic Study (SN-DREAMS, report 12). Diabetol Metab Syndr 2011. 3:9. [DOD] [CrossRef]
- Wu JC, Li XH, Peng YD, Wang JB, Tang JF, Wang YF. Association of two glyoxalase I gene polymorphisms with nephropathy and retinopathy in Type 2 diabetes. J Endocrinol Invest 2011. 34:e343-e348. [DOD]
- Wang Y, Ng MC, Lee SC, So WY, Tong PC, Cockram CS, Critchley JA, Chan JC. Phenotypic heterogeneity and associations of two aldose reductase gene polymorphisms with nephropathy and retinopathy in type 2 diabetes. Diabetes Care 2003. 26:2410-2415. [DOD] [CrossRef]
- Tian C, Fang S, Du X, Jia C. Association of the C47T polymorphism in SOD2 with diabetes mellitus and diabetic microvascular complications: a meta-analysis. Diabetologia 2011. 54:803-811. [DOD] [CrossRef]
- Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 2006. 114:597-605. [DOD] [CrossRef]
- Niu W, Qi Y, Wu Z, Liu Y, Zhu D, Jin W. A meta-analysis of receptor for advanced glycation end products gene: Four well-evaluated polymorphisms with diabetes mellitus. Mol Cell Endocrinol 2012. 358(1):9-17. [DOD] [CrossRef]
- Yuan D, Yuan D, Liu Q. Association of the receptor for advanced glycation end products gene polymorphisms with diabetic retinopathy in type 2 diabetes: a meta-analysis. Ophthalmologica 2012. In press. [DOD]
- Niedowicz DM, Daleke DL. The role of oxidative stress in diabetic complications. Cell Biochem Biophys 2005. 43:289-330. [DOD] [CrossRef]
- Cilensek I, Mankoc S, Petrovic MG, Petrovic D. GSTT1 null genotype is a risk factor for diabetic retinopathy in Caucasians with type 2 diabetes, whereas GSTM1 null genotype might confer protection against retinopathy. Dis Markers 2012. 32:93-99. [DOD]
- Toda N, Nakanishi-Toda M. Nitric oxide: ocular blood flow, glaucoma, and diabetic retinopathy. Prog Retin Eye Res 2007. 26:205-238. [DOD] [CrossRef]
- Cilensek I, Mankoc S, Petrovic MG, Petrovic D. The 4a/4a genotype of the VNTR polymorphism for endothelial nitric oxide synthase (eNOS) gene predicts risk for proliferative diabetic retinopathy in Slovenian patients (Caucasians) with type 2 diabetes mellitus. Mol Biol Rep 2012. In press. [DOD]
- Awata T, Inoue K, Kurihara S, Ohkubo T, Watanabe M, Inukai K, Inoue I, Katayama S. A common polymorphism in the 5'-untranslated region of the VEGF gene is associated with diabetic retinopathy in type 2 diabetes. Diabetes 2002. 51:1635-1639. [DOD] [CrossRef]
- Mirshahi A, Roohipoor R, Lashay A, Mohammadi SF, Abdoallahi A, Faghihi H. Bevacizumab-augmented retinal laser photocoagulation in proliferative diabetic retinopathy: a randomized double-masked clinical trial. Eur J Ophthalmol 2008. 18:263-269. [DOD]
- Gonzalez VH, Giuliari GP, Banda RM, Guel DA. Intravitreal injection of pegaptanib sodium for proliferative diabetic retinopathy. Br J Ophthalmol 2009. 93:1474-1478. [DOD] [CrossRef]
- Hsieh CH, Pei D, Hung YJ, Hsiao FC. Association between retinoid-X receptor-gamma genetic polymorphisms and diabetic retinopathy. Genet Mol Res 2011. 10(4):3545-3551. [DOD]
- Yang B, Millward A, Demaine A. Functional differences between the susceptibility Z-2/C-106 and protective Z+2/T-106 promoter region polymorphisms of the aldose reductase gene may account for the association with diabetic microvascular complications. Biochim Biophys Acta 2003. 1639:1-7. [DOD]
- Ng DP. Human genetics of diabetic retinopathy: current perspectives. J Ophthalmol 2010. 2010. [DOD]
- Sobrin L, Green T, Sim X, Jensen RA, Tai ES, Tay WT, Wang JJ, Mitchell P, Sandholm N, Liu Y, et al. Candidate gene association study for diabetic retinopathy in persons with type 2 diabetes: the Candidate gene Association Resource (CARe). Invest Ophthalmol Vis Sci 2011. 52:7593-7602. [DOD] [CrossRef]
- Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA 2002. 288:2579-2588. [DOD] [CrossRef]
- Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol 2010. 6:665-675. [DOD] [CrossRef]
- Zaidi SK, Young DW, Montecino M, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Architectural epigenetics: mitotic retention of mammalian transcriptional regulatory information. Mol Cell Biol 2010. 30:4758-4766. [DOD] [CrossRef]
- Stein GS, Zaidi SK, Stein JL, Lian JB, van Wijnen AJ, Montecino M, Young DW, Javed A, Pratap J, Choi JY, et al. Transcription-factor-mediated epigenetic control of cell fate and lineage commitment. Biochem Cell Biol 2009. 87:1-6. [DOD] [CrossRef]
- Chen S, Feng B, George B, Chakrabarti R, Chen M, Chakrabarti S. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am J Physiol Endocrinol Metab 2010. 298:E127-E137. [DOD] [CrossRef]
- Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, Calkin AC, Brownlee M, Cooper ME, El-Osta A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009. 58:1229-1236. [DOD] [CrossRef]
- Intensive blood-glucose control with sulphonylureas or insuln copared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study Group. Lancet 1998. 352(9131):837-853. [DOD] [CrossRef]
- Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008. 358:2560-2572. [DOD] [CrossRef]
- Chew EY, Ambrosius WT, Davis MD, Danis RP, Gangaputra S, Greven CM, Hubbard L, Esser BA, Lovato JF, Perdue LH, et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 2010. 363:233-244. [DOD] [CrossRef]
- Ismail-Beigi F, Craven T, Banerji MA, Basile J, Calles J, Cohen RM, Cuddihy R, Cushman WC, Genuth S, Grimm RH Jr, et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010. 376:419-430. [DOD] [CrossRef]
- Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, Zieve FJ, Marks J, Davis SN, Hayward R, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009. 360:129-139. [DOD] [CrossRef]
- Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH Jr, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008. 358:2545-2559. [DOD] [CrossRef]
- Gerstein HC, Miller ME, Genuth S, Ismail-Beigi F, Buse JB, Goff DC Jr, Probstfield JL, Cushman WC, Ginsberg HN, Bigger JT, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med 2011. 364:818-828. [DOD] [CrossRef]
This article has been cited by other articles:

|
Role of social interaction, exercise, diet, and age on developing and untreated diabetes in cynomolgus monkeys
Yue F, Zhang G, Quintero JE, Gash DM, Zhang Z
Exp Gerontol 2017. 96:82-88
|
|

|
Studies on glycoxidatively modified human IgG: Implications in immuno-pathology of type 2 diabetes mellitus
Islam S, Moinuddin, Mir AR, Arfat MY, Alam K, Ali A
Int J Biol Macromol 2017. 104(Pt A):19-29
|
|

|
Role of nutrients and mTOR signaling in the regulation of pancreatic progenitors development
Elghazi L, Blandino-Rosano M, Alejandro E, Cras-Meneur C, Bernal-Mizrachi E
Mol Metab 2017. 6(6):560-573
|
|

|
Trace element status in patients with type 2 diabetes in Norway: The HUNT3 Survey
Simic A, Hansen AF, Asvold BO, Romundstad PR, Midthjell K, Syversen T, Flaten TP
J Trace Elem Med Biol 2017. 41:91-98
|
|

|
ENPP1 121Q functional variant enhances susceptibility to coronary artery disease in South Indian patients with type 2 diabetes mellitus
Sumi S, Ramachandran S, RamanKutty V, Patel MM, Anand TN, Mullasari AS, Kartha CC
Mol Cell Biochem 2017. In press
|
|

|
Pulmonary Function and Retrobulbar Hemodynamics in Subjects With Type 2 Diabetes Mellitus
Tai H, Wang MY, Zhao YP, Li LB, Jiang XL, Dong Z, Lv XN, Liu J, Dong QY, Liu XG, Kuang JS
Respir Care 2017. 62(5):602-614
|
|

|
Herpes zoster as a presentation of diabetes mellitus
Verma VK, Ram VS, Singh PS, Kumar M, Awasthi S, Kela D
Int J Res Med Sci 2017. 5(5):1878-1881
|
|

|
Arsenic Exposure and Type 2 Diabetes: MicroRNAs as Mechanistic Links?
Beck R, Styblo M, Sethupathy P
Curr Diab Rep 2017. 17(3):18
|
|

|
Common variants of HNF1A gene are associated with diabetic retinopathy and poor glycemic control in normal-weight Japanese subjects with type 2 diabetes mellitus
Morita K, Saruwatari J, Tanaka T, Oniki K, Kajiwara A, Miyazaki H, Yoshida A, Jinnouchi H, Nakagawa K
J Diabetes Complications 2017. 31(2):483-488
|
|

|
The FOXO1 Gene-Obesity Interaction Increases the Risk of Type 2 Diabetes Mellitus in a Chinese Han Population
Gong L, Li R, Ren W, Wang Z, Wang Z, Yang M, Zhang S
J Korean Med Sci 2017. 32(2):264-271
|
|
|
Stress increases the risk of type 2 diabetes onset in women: A 12-year longitudinal study using causal modelling
Harris ML, Oldmeadow C, Hure A, Luu J, Loxton D, Attia J
Plos One 2017. 12(2):e0172126
|
|

|
Dietary salt intake and diabetes complications in patients with diabetes: An overview
Horikawa C, Sone H
J Gen Fam Med 2017. 18(1):16-20
|
|
|
Advances in epigenetic DNA methylation and diabetes mellitus
Hu ZW, Lu Q, Zhu LL
Genet Dis Stud 2017. 1(1):7-13
|
|
.jpg)
|
The effect of alogliptin on pulmonary function in obese patients with type 2 diabetes inadequately controlled by metformin monotherapy
Tai H, Wang MY, Zhao YP, Li LB, Dong QY, Liu XG, Kuang JS
Medicine (Baltimore) 2016. 95(33):e4541
|
|

|
Gliptins in managing diabetes - reviewing computational strategy
Sneha P, Doss CG
Life Sci 2016. 166:108-120
|
|

|
Genetic Investigation of Complement Pathway Genes in Type 2 Diabetic Retinopathy: An Inflammatory Perspective
Yang MM, Wang J, Ren H, Sun YD, Fan JJ, Teng Y, Li YB
Mediators Inflamm 2016. 2016:1313027
|
|

|
SUMO4 163 G>A variation is associated with kidney disease in Indian subjects with type 2 diabetes
Sinha N, Yadav AK, Kumar V, Dutta P, Bhansali A, Jha V
Mol Biol Rep 2016. 43(5):345-348
|
|
|
MicroRNA-194 Modulates Glucose Metabolism and Its Skeletal Muscle Expression Is Reduced in Diabetes
Latouche C, Natoli A, Reddy-Luthmoodoo M, Heywood SE, Armitage JA, Kingwell BA
Plos One 2016. 11(5):e0155108
|
|

|
A study on the association of diabetic dermopathy with nephropathy and retinopathy in patients with type 2 diabetes mellitus
Mirhoseini M, Saleh N, Momeni A, Deris F, Asadi-Samani M
J Nephropathol 2016. 5(4):139-143
|
|

|
Association of Urinary Phthalates with Self-Reported Eye Affliction/Retinopathy in Individuals with Diabetes: National Health and Nutrition Examination Survey, 2001-2010
Mamtani M, Curran JE, Blangero J, Kulkarni H
J Diabetes Res 2016. 2016:7269896
|
|

|
Type 2 diabetes mellitus disease risk genes identified by genome wide copy number variation scan in normal populations
Prabhanjan M, Suresh RV, Murthy MN, Ramachandra NB
Diabetes Res Clin Pract 2016. 113:160-170
|
|

|
Bariatric Surgery or Conventional Medical Therapy?: Which Is Best for Severely Obese Adults With Type 2 Diabetes?
Mayer J, Dwyer J
Nutr Today 2016. 51(5):233-241
|
|

|
Association between -308G/A TNFA Polymorphism and Susceptibility to Type 2 Diabetes Mellitus: A Systematic Review
Luna GI, da Silva IC, Sanchez MN
J Diabetes Res 2016. 2016:6309484
|
|

|
Gut Microbiota in Type 2 Diabetes Individuals and Correlation with Monocyte Chemoattractant Protein1 and Interferon Gamma from Patients Attending a Tertiary Care Centre in Chennai, India
Pushpanathan P, Srikanth P, Seshadri KG, Selvarajan S, Pitani RS, Kumar TD, Janarthanan R
Indian J Endocrinol Metab 2016. 20(4):523-530
|
|

|
Glucagon secretion from pancreatic alpha-cells
Briant L, Salehi A, Vergari E, Zhang Q, Rorsman P
Ups J Med Sci 2016. 121(2):113-119
|
|

|
Atorvastatin up-regulates the expression and activity of renal Cytochrome P450 3A2 in diabetic rats
Malekinejad H, Alizadeh-Fanaloub S, Hobbenaghic R, Rokhsartalb-Azarb S
J Appl Biomed 2016. 14(1):25-34
|
|
|
Inference of domain-disease associations from domain-protein, protein-disease and disease-disease relationships
Zhang W, Coba MP, Sun F
BMC Syst Biol 2016. 10(Suppl 1):4
|
|

|
Mean platelet volume is associated with myocardial perfusion defect in diabetic patients
Sahin S, Altunkas F, Karaman K, Sarikaya S, Akyol L, Borekci E, Yilmaz YK, Karacavus S, Erbay AR
South Afr J Diabetes Vasc Dis 2016. 13(2):77-80
|
|

|
Spousal Concordance of Diabetes Mellitus among Women in Ajman, United Arab Emirates
Al-Sharbatti SS, Abed YI, Al-Heety LM, Basha SA
Sult Qab Univ Med J 2016. 16(2):e197-e202
|
|

|
Metabolic and physiologic effects from consuming a hunter-gatherer (Paleolithic)-type diet in type 2 diabetes
Masharani U, Sherchan P, Schloetter M, Stratford S, Xiao A, Sebastian A, Nolte Kennedy M, Frassetto L
Eur J Clin Nutr 2015. 69(8):944-948
|
|

|
Association of adiponectin gene polymorphisms with an elevated risk of diabetic peripheral neuropathy in type 2 diabetes patients
Ji ZY, Li HF, Lei Y, Rao YW, Tan ZX, Liu HJ, Yao GD, Hou B, Sun ML
J Diabetes Complications 2015. 29(7):887-892
|
|

|
GADA and anti-ZnT8 complicate the outcome of phenotypic type 2 diabetes of adults
Haller-Kikkatalo K, Pruul K, Kisand K, Nemvalts V, Reimand K, Uibo R
Eur J Clin Invest 2015. 45(3):255-262
|
|

|
Higher titers of anti-Chlamydia pneumoniae IgG in diabetic retinopathy: a cross-sectional study
Banaee T, Daneshvar Kakhki R, Abrishami M, Mahmoudi M, Farzadnia M
Diabetes Metab Res Rev 2015. 31(2):168-174
|
|
|
An interpretable rule-based diagnostic classification of diabetic nephropathy among type 2 diabetes patients
Huang GM, Huang KY, Lee TY, Weng JT
BMC Bioinformatics 2015. 16(Suppl 1):S5
|
|
|
C-reactive protein genetic variant is associated with diabetic retinopathy in Chinese patients with type 2 diabetes
Peng D, Wang J, Zhang R, Tang S, Jiang F, Chen M, Yan J, Sun X, Wang T, Wang S, Bao Y, Hu C, Jia W
BMC Endocr Disord 2015. 15:8
|
|
|
Copy number variation: markers and predictors for type 2 diabetes
Ramirez-Valverde AG, Antunez-Ortiz DL, Mendez-Beleche A, Flores-Alfaro E, Ascencio-Montiel IJ, Cruz M
Rev Med Inst Mex Seguro Soc 2015. 53(3):348-355
|
|

|
Replication study of the association of rs7578597 in THADA, rs10886471 in GRK5, and rs7403531 in RASGRP1 with susceptibility to type 2 diabetes among a Japanese population
Sakai K, Imamura M, Tanaka Y, Iwata M, Hirose H, Kaku K, Maegawa H, Watada H, Tobe K, Kashiwagi A, Kawamori R, Maeda S
Diabetol Int 2015. 1:2190-1678
|
|

|
Neutrophil to Lymphocyte Ratio as an Indicator of Presence of Coronary Artery Disease in Diabetic Patients
Fernando ML, Silambanan S, Malar J
Int J Clin Biochem Res 2015. 2(3):143-147
|
|

|
Long-term changes in dietary and food intake behaviour in the Diabetes Prevention Program Outcomes Study
Jaacks LM, Ma Y, Davis N, Delahanty LM, Mayer-Davis EJ, Franks PW, Brown-Friday J, Isonaga M, Kriska AM, Venditti EM, Wylie-Rosett J, Diabetes Prevention Program Research Group
Diabet Med 2014. 31(12):1631-1642
|
|

|
Genetic determinants of atherosclerosis, obesity, and energy balance in consomic mice
Spiezio SH, Amon LM, McMillen TS, Vick CM, Houston BA, Caldwell M, Ogimoto K, Morton GJ, Kirk EA, Schwartz MW, Nadeau JH, LeBoeuf RC
Mamm Genome 2014. 25(11-12):549-563
|
|

|
Role of BKCa channels in diabetic vascular complications
Qian L, Liu X, Wang R
Chin Med J (Engl) 2014. 127(9):1775-1781
|
|
|
Effects of swimming training on endothelium-dependent vasodilation function of aorta in diabetic rats and its mechanisms
Yang H, Wang Y, Li W, Qian J
Chin J Rehabil Med 2014. 2014(8):740-744
|
|

|
Update on health literacy and diabetes
Bailey SC, Brega AG, Crutchfield TM, Elasy T, Herr H, Kaphingst K, Karter AJ, Moreland-Russell S, Osborn CY, Pignone M, Rothman R, Schillinger D
Diabetes Educ 2014. 40(5):581-604
|
|

|
The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes
Martin SD, McGee SL
Biochim Biophys Acta 2014. 1840(4):1303-1312
|
|

|
Mean platelet volume is associated with myocardial perfusion defect in diabetic patients
Sarikaya S, Sahin S, Akyol L, Borekci E, Yilmaz YK, Altunkas F, Karaman K, Karacavus S, Erbay AR
Cardiovasc J Afr 2014. 25(3):110-113
|
|

|
Insulin resistance, type 1 and type 2 diabetes, and related complications: current status and future perspective
Ndisang JF, Rastogi S, Vannacci A
J Diabetes Res 2014. 2014:276475
|
|

|
Type 2 Diabetes Mellitus: The Concerned Complications and Target Organs
Kumara A, Bhartib SK, Kumar A
Apollo Med 2014. 11(3):161-166
|
|

|
10-year incidence of diabetes and associated risk factors in Greece: the ATTICA study (2002-2012)
Koloverou E, Panagiotakos DB, Pitsavos C, Chrysohoou C, Georgousopoulou EN, Pitaraki E, Metaxa V, Stefanadis C, ATTICA Study Group
Rev Diabet Stud 2014. 11(2):181-189
|
|

|
Of rodents and men: species-specific glucose regulation and type 2 diabetes research
Chandrasekera PC, Pippin JJ
ALTEX 2014. 31(2):157-176
|
|
|
Association between GSTT1 and GSTM1 gene polymorphisms and Type II diabetes mellitus patients in Basra - Iraq
Al-Badran AI, Al-Mayah MK
Int J Curr Microbiol App Sci 2014. 3(11):288-299
|
|
|
Predictive factors of diabetic complications: a possible link between family history of diabetes and diabetic retinopathy
Maghbooli Z, Pasalar P, Keshtkar A, Farzadfar F, Larijani B
J Diabetes Metab Disord 2014. 13:55
|
|

|
Landscape of the relationship between type 2 diabetes and coronary heart disease through an integrated gene network analysis
Dong C, Tang L, Liu Z, Bu S, Liu Q, Wang Q, Mai Y, Wang DW, Duan S
Gene 2014. 539(1):30-36
|
|

|
Common polymorphism in the cannabinoid type 1 receptor gene (CNR1) is associated with microvascular complications in type 2 diabetes
Buraczynska M, Wacinski P, Zukowski P, Dragan M, Ksiazek A
J Diabetes Complications 2014. 28(1):35-39
|
|
|
Meta-analysis identifies gene-by-environment interactions as demonstrated in a study of 4,965 mice
Kang EY, Han B, Furlotte N, Joo JW, Shih D, Davis RC, Lusis AJ, Eskin E
PLoS Genet 2014. 10(1):e1004022
|
|

|
Diabetes Mellitus Type 2 Prevalence, Risk Factors and Complications in the Region of Kardzhali, Bulgaria
Staykova J, Manolova A, Tzolova G
J Pharm Pharmacol 2014. 2:145-151
|
|

|
Association of CFH and CFB gene polymorphisms with retinopathy in type 2 diabetic patients
Wang J, Yang MM, Li YB, Liu GD, Teng Y, Liu XM
Mediators Inflamm 2013. 2013:748435
|
|
|
Gastric inhibitory polypeptide receptor methylation in newly diagnosed, drug-naïve patients with type 2 diabetes: a case-control study
Canivell S, Ruano EG, Siso-Almirall A, Kostov B, Gonzalez-de Paz L, Fernandez-Rebollo E, Hanzu F, Parrizas M, Novials A, Gomis R
Plos One 2013. 8(9):e75474
|
|
|
Intracellular adhesion molecule-1 K469E gene polymorphism and risk of diabetic microvascular complications: a meta-analysis
Su X, Chen X, Liu L, Chang X, Yu X, Sun K
Plos One 2013. 8(7):e69940
|
|

|
CIDE - A gene expression in patients with abdominal obesity and LDL hyperlipoproteinemia qualified for surgical revascularization in chronic limb ischemia
Feldo M, Kocki J, Lukasik S, Bogucki J, Feldo J, Terlecki P, Kesik J, Wronski J, Zubilewicz T
Pol Przegl Chir 2013. 85(11):644-648
|
|

|
Evaluation of Neutrophil to Lymphocyte ratio as an Indicator of Presence of Coronary Artery Disease in Diabetic Patients
Sahin S, Sarikaya S, Akyol L, Altunkas F, Karaman K
Natl J Med Res 2013. 3(4):300-303
|
|

|
The diabetic lung - a new target organ?
Pitocco D, Fuso L, Conte EG, Zaccardi F, Condoluci C, Scavone G, Incalzi RA, Ghirlanda G
Rev Diabet Stud 2012. 9(1):23-35
|
|
|


)
)
)

