Review
| Rev Diabet Stud,
2012,
9(2-3):68-81 |
DOI 10.1900/RDS.2012.9.68 |
Immunotherapy in Autoimmune Type 1 Diabetes
Benno Weigmann1, Randi K. Franke2, Carolin Daniel2
1Research Campus of the Friedrich-Alexander University Erlangen-Nuernberg, Medical Clinic I, 91052 Erlangen, Germany
2Helmholtz Zentrum Muenchen - German Center on Environmental Health, Institute of Diabetes Research 1, Junior Group Immunological Tolerance in Type 1 Diabetes, Ingolstaedter Landstrasse 1, Neuherberg, 85764 Munich, Germany
Address correspondence to: Carolin Daniel, e-mail: carolin.daniel@helmholtz-muenchen.de
Manuscript submitted October 18, 2012; resubmitted November 3, 2012; accepted November 7, 2012.
Keywords: Immunotherapy, autoimmunity, type 1 diabetes, insulin, regulatory T cells, Foxp3, mimetope, antigen, tolerance, suppression
Abstract
Type 1 diabetes (T1D) is a chronic autoimmune disease affecting millions of people worldwide. The disease is characterized by the loss of self-tolerance to the insulin-producing β-cells in the pancreas, the destruction of β-cells, and finally the development of chronic hyperglycemia at diagnosis of T1D. Its incidence and prevalence are rising dramatically, highlighting the need for immunotherapeutic strategies able to prevent or treat the disease in a safe and specific manner. Immunotherapeutic strategies are being developed, and aim to restore immunological self-tolerance, thereby limiting unwanted immunity and β-cell destruction. Foxp3+ regulatory T (Treg) cells exert essential functions to maintain and restore immunological self-tolerance. The identification of the transcription factor Foxp3 as the specification factor for the Treg cell lineage facilitated our understanding in the biology of Treg generation and function. This review highlights the current understanding of immunotherapeutic approaches as preventative and curative measures for autoimmune T1D. It includes an overview on early immunointervention studies, which made use of general immunosuppressive agents such as cyclosporin A, followed by a discussion on newly emerging clinical trials. Besides non-antigen-specific therapies, particular attention is given to antigen-specific generation of Foxp3+ Treg cells and their potential use to limit autoimmunity such as T1D.
Abbreviations: AIDA - anti-interleukin-1 in diabetes action; CsA - cyclosporin A; CTLA4 - cytotoxic T lymphocyte antigen 4; DPT - Diabetes Prevention Trial; FoxP3 - forkhead box P3 protein (also known as scurfin); GAD65 - glutamic acid decarboxylase 65; GWAS - genome-wide association studies; HLA - human leukocyte antigen; IAA - insulin autoantibody; IA-2 - insulinoma antigen; Ig - immunoglobulin; IL-1β - interleukin-1β; IMPDH - inosine monophosphate dehydrogenase; IPEX - immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome; MHC - major histocompatibility complex; MMF - mycophenolate mofetil; MPA - mycophenolic acid; mTOR - mammalian target of rapamycin; NOD - non-obese diabetic; OKT3 - orthoclone muromonab CD3; PI3K - phosphatidylinositide 3-kinase; Rag - recombination activating gene; SCID - severe combined immunodeficiency (mouse model without functional T and B cells); T1D - type 1 diabetes; TCR - T cell receptor; TGFβ - transforming growth factor beta; TNF - tumor necrosis factor; Treg - regulatory T (cell); VNTR - variable number tandem repeat; ZnT8 - zinc transporter 8
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disorder characterized by specific immune destruction of the insulin-producing pancreatic β-cells [1]. The inaccessibility of the human pancreas has led to relatively limited studies of its role in the biological mechanisms of T1D. Despite this limitation, several key studies have provided evidence for the infiltration of immune cells in the pancreatic islets (insulitis) before and at diagnosis of the disease [1]. Autoreactive T cells and other mononuclear cells infiltrate the islets initiating and maintaining insulitis, which ultimately leads to β-cell death, diabetes, and a lifelong requirement for insulin therapy [1]. However, detailed understanding of the precise mechanisms dictating the immunological events that control autoimmune destruction is still missing and needs to be elucidated.
The incidence of T1D is rapidly rising in children, with a predicted 70% increase in incidence over the next 15 years in Europe. Furthermore, the age of onset is also decreasing, with a predicted doubling of cases in children under the age of 5 years during the same period, which highlights the dramatic impact of T1D on future public health.
Despite refined treatment with insulin therapy, long-term complications, including nephropathy, retinopathy, neuropathy, and cardiovascular disease, can appear [2, 3]. It is believed that T1D develops as a result of genetic predisposition and unknown environmental factors. Genetic susceptibility to T1D is largely conferred by the inheritance of the human leukocyte antigen (HLA) class II haplotypes of HLA-DR and HLA-DQ located within the major histocompatibility complex (MHC) on chromosome 6p21 [4]. Genome-wide association studies (GWAS) have revealed that 25 non-HLA-associated loci contribute additional risk to autoimmune T1D. The vast majority of these genes encode proteins that are involved in immune function and regulation [5, 6].
Recent longitudinal studies of a large cohort of monozygotic twins, initially discordant for T1D, demonstrated that 65% of twins ultimately develop T1D over a follow-up period of 43 years. These data support the suggestion that all identical twins will eventually become concordant providing they live sufficiently long [7]. The clinical definition of diabetes is determined by the appearance of glucose levels at which end-organ damage is known to occur. It does not account for the fact that the emergence of autoimmunity predates the diagnosis by several years [8].
The large number of identified genes supports the view that several signaling pathways are involved in the development of autoimmunity, finally resulting in the loss of tolerance to pancreatic islets in humans [9]. These findings have important implications for the development of immunomodulatory strategies for T1D. Since the disease is dependent on the individual, not all approaches are equally effective in all subjects. The chronic autoimmune process that usually precedes the development of clinical T1D can be monitored by autoantibody responses. Autoantibodies per se are not directly pathogenic, but they represent biomarkers for the development and status of the autoimmune disease [10]. It has been observed that individuals expressing specific autoantibodies reacting with 2 of 4 well-defined autoantigens (insulin, glutamic acid decarboxylase (GAD65), insulinoma antigen (IA-2), and islet zinc transporter (ZnT8)) eventually progress to the development of T1D [6].
Insulin-specific antibodies may be detected in some children as young as 6-12 months of age and may predate the development of T1D by up to 10 years [11]. The age of development of autoimmunity in T1D is related to the burden of susceptibility genes, with individuals at high genetic risk developing autoimmunity before the age of 5 years [12]. In line with this view, it became clear that young children can progress rapidly, within months after the appearance of autoantibodies, to overt diabetes. In contrast, some children take more than a decade to develop the disease, with evidence of chronic beta-cell destruction preceding diabetes over years, as discussed above [6]. Several studies have indicated that the appearance of autoantibodies is acute, and during the prediabetic period different autoantibodies rise and fall, but usually more than two are expressed, sometimes for decades. Similarly, in the setting of the non-obese diabetic (NOD) mouse model, the development of insulin autoantibodies occurs at an early age of around 15 days after birth. However, the characterization of other autoantibodies has met with challenges when measured with highly specific assays [1]. Alternatively, the characterization of T-cell-mediated autoimmunity to multiple autoantigens is more advanced in the NOD mouse model than in human T1D.
After the critical contribution of MHC in defining the risk of developing T1D, the second-most important locus determining T1D is the insulin gene. It has become evident that a polymorphism of a variable number tandem repeat (VNTR) 50 of the insulin gene, and not a variation in the insulin gene sequence itself, contributes to the risk of developing T1D. The long variant of the VNTR is associated with both greater insulin message intrathymically and protection from the development of T1D [13, 14]. Insulin autoantibodies are usually, but not always, the first autoantibodies to appear in young children developing T1D [15]. Insulin autoantibody levels were found to inversely correlate with the age at which diabetes develops [6]. When detected at first, insulin autoantibodies are of high affinity and highly predictive. Insulin works as a critical target of both B cells and T cells in human T1D and in NOD mice [16-18]. In line with this observation, mice which express a mutant insulin gene product not recognized by T cells do not develop the disease [19]. In the context of human T1D, the extent of intrathymic insulin expression has been linked to the incidence of diabetes [13]. In mice and humans, the T cell response to insulin is highly focused on a segment of the B-chain encompassing residues 9-23 [16-18], and the human epitope is identical to that of the murine insulin.
It has been shown that the removal of insulin 1, which is mainly expressed in the islets, is associated with a blockade of disease progression [20]. Conversely, removal of the insulin 2 gene greatly promotes disease progression, most likely because of a deficiency in thymic insulin expression and subsequent loss of central tolerance [20-22]. Finally, retroviral introduction of T cell receptor (TCR) genes from insulin-reactive CD4+ T cells into SCID NOD mice is sufficient to support the development of insulin autoimmunity in a significant proportion of mice [23].
Based on considerable evidence for the importance of insulin as an autoantigen in NOD and human T1D, and combined with the fact that in humans T1D incidence is rising dramatically, especially in young children, intensive efforts have been made to develop self-antigen-specific (e.g. insulin-specific) immunotherapies, with the ultimate goal of achieving a safe and specific prevention of T1D. Indeed, some advances have been made in the development of self-antigen-specific immunotherapy. However, translating these strategies from bench to bedside has met with challenges. Also, the success of clinical trials using natural self-antigens for induction of self-tolerance, e.g. natural insulin in T1D, has been limited. Thus far, three trials of secondary immunoprevention in T1D using whole insulin have been completed:
1. DPT-1 (oral [24] and parental [25])
2. Intranasal Insulin Trial I [26]
3. Intranasal Insulin for Prevention of T1D trial [27]
These studies using oral or intranasal insulin therapy in humans have had either no or limited clinical benefit in human T1D [28, 29]. Subsequent subgroup analysis of data from the DPT-1 trial supported the hypothesis that patients with high insulin-specific antibody titres who received oral insulin had a delay in progression to T1D [24]. The beneficial side-effect profile of oral and inhaled insulin in secondary immuneprevention trials has led to the establishment of a Phase I trial (named Pre-POINT) to assess the effects of mucosal insulin therapy for primary immunoprevention [30].
However, thus far the applied immunomodulatory approaches did not address the question of whether the choice of antigen, the time point and route of administration were suitable to induce tolerance in the respective population at risk of developing T1D. Furthermore, no specific protocols for the conversion of naïve T cells into Foxp3-expressing regulatory T (Treg) cells have been explored. Therefore, it has been proposed that disease state, antigen dose, route of administration, study cohort, and the choice of antigen, e.g. insulin vs. insulin B chain peptides, work as critical parameters in inducing tolerance [28, 29]. Moreover, an improved understanding of the requisites for an efficient induction of human self-antigen-specific Foxp3+ Treg cells in an autoimmune setting such as T1D is needed.
When developing a novel T1D immunotherapeutic approach, it must build on known approaches for manipulating autoimmune mechanisms to devise novel therapeutic strategies that address these unmet medical requirements, as outlined above. In the context of the young patient population increasingly affected by T1D, the challenge is to achieve clinical efficacy in the absence of chronic immunosuppression to avoid compromising the host’s defense against infections and tumors. Ultimately, the scientific efforts should result in a range of immunotherapeutic options that combine short-term β-cell preservation with long-term modulation of autoimmunity by the restoration of immunological self-tolerance. As in other immune-mediated diseases, it is unlikely that a single treatment will be effective in all patients. Biomarkers indicating clinical and immunologic efficacy will be required, along with the identification of combinatorial approaches that can guide immunotherapy for individual patients. In the present review, we discuss the requisites and opportunities for immunotherapeutic strategies to interfere with autoimmunity such as T1D, with particular emphasis on the generation of self-antigen specific Foxp3+ Treg cells.
Non-antigen-specific immunomodulative approaches
Based on the autoimmune etiology of T1D, early intervention strategies employed the use of immunosuppressive agents to interfere with the development of an autoreactive immune response. Some of these approaches were successful in inducing and prolonging clinical remission. However, protective effects were lost upon drug removal since their effect relied on general immunosuppression and was not based on long-term induction of tolerance. Moreover, the application of these drugs was frequently accompanied by deleterious side effects, limiting their broad application for prolonged time periods.
Cyclosporin A
One of the earliest compounds applied as an immunosuppressing agent in patients with T1D was cyclosporin A (CsA). CsA is an 11-amino acid cyclic peptide of fungal origin with a strong immunosuppressive capacity. It is a calcineurin inhibitor that interferes with TCR-mediated signal transduction, thereby inhibiting T cell activation, and the production of interleukin 2 (IL-2) by T cells, which is then able to limit the amplification of immune responses [31]. In T1D, CsA provided the first proof of concept that T-cell-directed immunosuppression was capable of preserving β-cell function and insulin production. Based on results from pilot studies [32, 33], two randomized phase II placebo-controlled studies were conducted [34, 35]. Based on these studies, it was concluded that treatment with CsA had no long-lasting effect on the course of T1D, persisting upon removal of compound administration [34, 36]. The need for chronic drug administration, the potential renal and pancreatic β-cell toxicity, and the cost for the drug led to the consensus that the risks outweighed the benefits, and the approach was dropped.
Mycophenolate mofetil
Mycophenolate mofetil (MMF) is an immunosuppressive drug which has been applied in the context of organ allograft rejection. It became clear that MMF possesses significant cytostatic effects on lymphocytes [37]. MMF functions as a pro-drug of mycophenolic acid (MPA), which is an inhibitor of inosine monophosphate dehydrogenase (IMPDH). IMPDH is the rate-limiting enzyme in the de novo synthesis of guanosine nucleotides, thereby playing a critical role in controlling the proliferation of T and B cells [38]. However, despite its efficacy in organ transplantation, a combination of MMF and a monoclonal antibody (daclizumab) targeting CD25, the alpha chain of the IL-2 receptor, which is widely used in transplantation [39], did not preserve β-cell function in newly diagnosed patients with T1D [40]. Also, there was no decrease in insulin requirement or improvement in metabolic control. One-third of patients treated with the combination of daclizumab and MMF suffered from serious adverse events. Although negative, these data are relevant as they highlight that it is not just any immunosuppressive regimen that can effectively treat T1D. The failure may be explained by the consideration that, by targeting CD4+CD25+ regulatory T cells (Tregs), daclizumab removes a cell subset from the immune system that plays an essential role in the maintenance of self-tolerance in T1D [41, 42].
Anti-CD20: rituximab
Rituximab is a chimeric antibody that targets the CD20 transmembrane receptor, which is expressed on all immature and mature B cells. Initially, rituximab was used for the treatment of non-Hodgkin's B cell lymphoma [43]. With respect to autoimmunity, rituximab has been combined with anti-proliferative agents to treat systemic lupus erythematosis and rheumatoid arthritis. However, these studies showed that both diseases present with relapses upon withdrawal of drug application. This result supports the concept that the effects are immunosuppressive but do not induce long-term tolerance [44, 45].
In a phase II clinical trial, rituximab was tested in patients with recent onset T1D. It became clear from these experiments that the treatment effect of rituximab was most prevalent within the first 3 months of application. Over this time period, the treatment was able to reduce the loss of C-peptide and insulin requirements. Later on, analyses revealed that the effects on C-peptide responses did not prevail. There was no statistically significant difference between patients that had received rituximab and the placebo-treated groups [46].
Cytotoxic T-lymphocyte-associated protein 4 immunoglobulin (CTLA4-Ig): abatacept and belatacept
CTLA4-Ig is a fusion protein consisting of the extracellular domain of CTLA4 and the Fc domain of an IgG1 antibody [47, 48]. It is well established that CTLA4 is expressed by activated CD8+ T cells. However, CTLA4 exerts its main function as a negative costimulatory molecule leading to the inhibition of helper T cell activity and enhancement of Treg immunosuppression. Its ligands comprise CD80 and CD86, which bind to CD28, thereby delivering the costimulatory signal needed for T cell activation. Moreover, CTLA4 is a target gene of Foxp3. It was shown that a Treg-cell-specific CTLA4 knockout or blockade is able to inhibit the ability of Tregs to control autoimmune reactions and anti-tumor immunity.
In the NOD mouse model of T1D, the application of CTLA-Ig showed conflicting results with respect to the progression of diabetes, while in some settings the administration of murine CTLA4-Ig worsened the development of diabetes [49-51]. In humans, CTLA4-Ig (abatacept) has been successfully used to treat psoriasis and rheumatoid arthritis [51]. In rheumatoid arthritis, a combined application of abatacept and methotrexate was appropriate to treat patients who did not respond to anti-tumor necrosis factor (TNF) agents. Abatacept has no tolerogenic characteristics and monthly infusions are sufficient to maintain immunosuppressive properties [53-56]. Application of abatacept was tested in a multicenter, double-blind, randomized controlled trial with recent onset diabetes patients. The drug was applied at doses of 10 mg/kg on days 1, 14, and 28, followed by monthly injections for a total of two years [57]. Abatacept treatment resulted in an estimated delay in C-peptide reduction of about 10 months. A longer follow-up is necessary to determine whether there is a persisting treatment effect maintained after cessation of application.
Anti-TNF therapy
Anti-TNF therapy has been established for the treatment of chronic pro-inflammatory autoimmune diseases such as rheumatoid arthritis and Crohn's disease. In NOD mice, the effect of TNF blockade varies depending on the age at which treatment is applied. The development of autoreactive T cells was demonstrated to be modulated by TNF treatment, suggesting an effect on the development of the intrathymic autoimmune repertoire [58-60]. However, the critical factors, which delineated the timing of the TNF effects, are far from being understood in detail. Therefore, translation of these findings to the clinic remains a challenge.
Treatment with a soluble recombinant TNF receptor fusion protein that binds TNF (etanercept) did not prevent T1D in humans [61, 62]. In a double-blind, placebo-controlled pilot study, 18 children with new-onset T1D were included and patients received placebo or etanercept twice a week [63]. After 24 weeks, a clear reduction in the required insulin dose could be observed in the etanercept group. No serious adverse effects were seen in this group. Further studies in a larger set are required to confirm this promising result.
Anti-interleukin-1 (IL 1) therapy: anakinra
Anti-IL 1 therapy has primarily been used for the treatment of rheumatoid arthritis [64]. Results from studies using models of T1D support the concept that anakinra exerted direct protective effects on β-cells, rather than on insulin resistance, which led to the observed improvement in metabolic control [65]. The improvement in C-peptide responses persisted for up to 39 weeks after cessation of treatment [66]. It is hypothesized that the positive effects from anakinra result from the blockade of pro-inflammatory signals from immune cells and islet cells, thereby limiting the IL-1β-mediated induction of β-cell death.
Preliminary studies in NOD mice suggest that anti-IL 1 application is able to lower the incidence of T1D. Results from IL-1 receptor (IL-1R) deficient NOD mice showed a delay, but not protection, from T1D development. This observation supports the view that anakinra will probably not be sufficient as a single agent to achieve full treatment success [67]. The anti-interleukin-1 in diabetes action (AIDA) study is about to test feasibility, safety, and efficacy of anti-IL-1 therapy in maintaining and/or enhancing β-cell function in people with new-onset T1D [68].
Canakinumab (anti-interleukin-1β)
Canakinumab is a fully human anti-interleukin-1β (anti-IL-1β) monoclonal antibody (IgG-1 class). Canakinumab is designed to bind to human IL-1β resulting in the functional neutralization of this proinflammatory cytokine. In a recent trial, repeated injections of canakinumab were assessed for their ability to preserve β-cell function in patients with recent onset T1D.
Anti-CD3 therapy: teplizumab and otelizumab
Studies in mouse models of T1D were supportive of the concept that the application of CD3-specific mAbs is able to promote tolerance induction to β-cell antigens in the pancreas [69]. OKT3, also called muromonab-CD3, was the first generation of antibodies developed for the prevention of solid organ transplant rejection [70, 71]. However, its application was associated with a severe cytokine release syndrome, which resulted from crosslinking and activation of T cell receptors by the binding of the murine antibody Fc portion to Fc receptors on human cells [72]. To limit the side effects caused by the mitogenic potential of mAbs, humanized CD3-specific Fc mutated mAbs were developed. Teplizumab, also called hOKT3γl, a FcR-non-binding, and otelixizumab (chAglyCD3), an aglycosylated FcR non-binding CD3-specific Ab, were tested in clinical trials on patients with recent-onset T1D. They demonstrated significantly reduced side-effects [73, 74]. Based on preclinical studies in the NOD mouse setting, two phase II trials―one of which was placebo controlled―were performed and showed a clear effect, with best success seen in patients with higher functional β-cell mass before the start of otelixizumab treatment [75-77]. Following up on these findings, phase III clinical trials (otelixumab, tolerx, GlaxoSmithKline trial, and teplizumab, MacroGenics/Eli Lilly trial) were performed. However, the design of these studies differed significantly from those of the completed phase II clinical trials. Both studies failed to meet their primary end point at 1 year. Importantly, when a post-hoc analysis of the data of the teplizumab study was performed, using the conventional end points by previous studies (C-peptide production and insulin needs), a clear treatment effect became apparent [78].
Self-antigen-specific immunomodulative strategies
Foxp3+ regulatory T (Treg) cells
Proper functioning of the immune system implies a tightly balanced regulation of effector mechanisms to avoid immune pathologies and autoimmunity. A variety of control mechanisms have been identified, including negative feedback circuits, which impact on activation, survival, or functioning of effector cells [79-82]. Treg cells, which can suppress self-reactive responses, are a vital and essential component in the maintenance of immunological self-tolerance [83, 84]. Early studies identified Treg cells as CD4+CD25+ T cells [85]. The identification of the X-chromosome-encoded transcription factor Foxp3 permitted a clearer understanding of Treg cell biology [86-88]. The pivotal impact of Foxp3+ Treg cells in the maintenance of immunological self-tolerance was further highlighted by the fatal autoimmune syndrome found in patients with immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, which develops in humans harboring mutations of the Foxp3 gene [89]. Likewise, scurfy mice, which lack a functional Foxp3+ protein because of a natural mutation, suffer from various spontaneous and early-onset organ-specific autoimmune diseases due to hyperactivation of CD4+ T cells [90]. It became clear from Treg adoptive transfer experiments in scurfy mice that Treg cells are essential in the avoidance of autoimmunity in mice. Moreover, enforced continuous expression of Foxp3 in conventional CD4+ T cells was able to confer a phenotype and function resembling Treg cells. In turn, conditional deletion of the Foxp3 gene reprograms Treg cells into pathogenic T cells [91, 92]. These results supported the view that Foxp3 functions as a specific marker of Treg cells, and acts as their lineage specification factor or master regulator.
Extrathymic differentiation of Foxp3 expressing Treg cells
Recognition of agonist TCR ligand works as a key element for the initiation of Treg cell generation, a developmental program which can run intra- [93, 94] or extrathymically [95-100]. Studies on extrathymic Treg cell generation in the peripheral adult immune system can have an important therapeutic potential. Our group has carefully established protocols for antigen-specific Treg conversion. It became clear that extrathymic conversion of naïve CD4+ T cells into Foxp3+-expressing Treg cells can be achieved in vivo by the delivery of strong-agonist TCR ligands under subimmunogenic conditions [95-100] (Figure 1). The concept of de novo induction of Foxp3+ Treg cells, rather than the expansion of already committed Treg cells, was further supported by the fact that Foxp3+ Tregs were absent in Rag-/- TCR-transgenic animals expressing only one particular TCR in the absence of a co-expressed TCR-agonist ligand. Cellular proliferation was found to function as a limiting factor for Treg conversion since the best conversion is seen in T cells that undergo only limited proliferation. Whereas, higher doses of TCR agonists induce robust proliferation and diminished conversion into Treg cells [99]. Recent data support the concept that such high doses of TCR ligands result in an activation of the PI3K/Akt/mTOR pathway [101], which could interfere with extrathymic Foxp3 induction [102]. Early blockade of PI3K signaling through the use of PI3K-mTOR inhibitors was found to promote Treg cell induction in vitro [98, 103] and in vivo [98]. In accordance with this concept, sustained Akt activation inhibited the stable induction of Foxp3 in peripheral Foxp3-CD4+ T cells [104].
The induction of Foxp3 upon subimmunogenic antigen exposure in vivo requires TGFβ receptor signaling and is inversely correlated with cellular proliferation, as mentioned above [99]. Extrathymically induced Foxp3+ Treg cells can prospectively be generated for suppression of unwanted immune responses since it has become clear that they are stable and independent of further antigen supply used to generate these cells [105, 106]. Recent studies support the concept that the extrathymically induced Foxp3+ Treg cells are stable upon subimmunogenic antigen delivery [106], and maintain their function even in an immunogenic context [107]. Re-encounter of antigen under immunogenic conditions does not cause loss of Treg cells or loss of their activity [105], but permits the expansion of Treg cells [99].
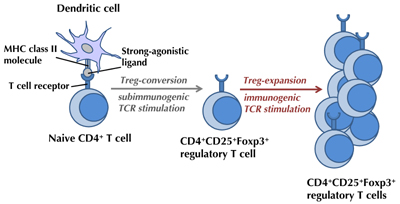 |
 |
Figure 1. Generation of extrathymic regulatory T (Treg) cells. Efficient in vivo generation of Foxp3+ Treg cells in the mouse system requires delivery of strong-agonistic T cell receptor (TCR)-ligands under subimmunogenic conditions and avoidance of activation of antigen-presenting and T cells. Immunogenic antigen stimulation is able to expand Treg cells once converted. |
|
Extrathymic induction of Treg cells in autoimmune T1D
In models unrelated to autoimmune disease, it was demonstrated that naïve CD4+ T cells can be extrathymically converted into Foxp3+ Treg cells in vivo if they are exposed to strong-agonistic antigens under subimmunogenic conditions [94-99]. Such extrathymic Treg cell induction strategies were used to prospectively generate transplantation tolerance in female mice to various male tissues [100]. However, initial studies investigating the feasibility of prospective extrathymic Foxp3 Treg generation in the prevention of the development of T1D by subimmunogenic delivery of natural insulin B chain epitopes in the NOD mouse showed only a short delay of disease progression.
As a possible scenario, it is believed that T1D and other autoimmune diseases develop when T cells with specificity for weakly binding TCR agonists, which may include self-antigens, escape thymic negative selection and evade into the periphery where they cause an autoreactive process [96, 108-112]. Specific modes of presentation and recognition have been proposed for certain self-antigens in the course of autoimmunity [108-116]. Also, higher epitope abundance in the respective organ (e.g. pancreatic islets) and local differences in peptide processing/truncating and/or presentation could possibly facilitate the activation of these self-reactive T cells in the periphery [114, 118].
It became clear from various studies that in autoimmune T1D insulin has a critical role as a self-antigen [19, 119]. As discussed above, in mice and humans, the T cell response to insulin is focused on the B-chain residues 9-23. In NOD mice, characterizing the relevant epitope(s) for the T cells within the insulin B:9-23 peptide has been a challenge. This difficulty underlines the significance of the concept that the insulin B-chain peptide may bind to the murine MHC class II molecule I-Ag7, and may be recognized by T cells in several overlapping binding registers [17, 113, 114, 120-122].
In a model proposed by John Kappler, the insulin B chain peptide was presented by I-Ag7 molecules in an unfavored low-affinity binding register, which resulted in weak agonistic activity of the peptide-MHC complex. The poor binding to I-Ag7 molecules was the result of an incompatibility between the p9 amino acid of the insulin epitope (arginine = R) and the particular I-Ag7 p9 pocket polymorphisms, which are highly linked with susceptibility to T1D [113, 114]. In this unfavored binding register, an arginine residue (=R) of the insulin epitope met another arginine (=R) in the positively charged p9 pocket of I-Ag7, thereby generating a highly unfavorable match [113, 114, 123, 124]. While these data represent a compelling model, it is challenged by other investigators who propose that binding of the insulin epitope to I-Ag7 largely takes place in other low-affinity registers [122].
In human T1D, the susceptibility MHC class II allele, HLA-DQ8, shares very similar binding pockets for peptide presentation with the homologous molecule in NOD mice, I-Ag7 [125]. The unique feature, which confers disease susceptibility in NOD mice, was equally identified in the peptide binding groove of human HLA-DQ8. It became clear that the polymorphism, which affects the presentation of insulin in NOD mice, i.e. the lack of one specific amino acid in the B chain of the MHC molecule, was identical in human HLA-DQ8 and I-Ag7 of NOD mice [123, 125]. Therefore, both may exhibit similar peptide binding interactions and similar modes of antigen presentation.
We showed that upon subimmunogenic application of a strong-agonistic insulin B-chain variant to young NOD mice, efficient insulin-specific conversion of naïve T cells into Foxp3+ Treg cells could be achieved. This approach has the ability to prevent the development of T1D [97]. In contrast to the natural insulin epitope, subimmunogenic Treg conversion with the strong-agonist mimetope resulted in high numbers of stable Foxp3+ Treg cells. These findings are supportive of the concept that low doses of strong-agonistic antigens are able to induce stable Foxp3+ Treg cells with high efficacy. Whereas, even high doses of poorly agonistic ligands fail to generate stable Foxp3+ Treg cells. These results indicate that ligand density cannot compensate for diminished agonistic activity in determining the efficacy and stability of induced Foxp3+ Treg cells [97, 126].
We studied the development of diabetes in NOD mice as a function of insulin autoantibody (IAA) indices at a young age before Treg-induction. We observed that autoantibody indices present in mice at 4 weeks of age impact the development of T1D in these mice. A correlation became apparent in that mice with higher IAA indices developed T1D earlier. We showed that the natural insulin B chain epitope conferred only limited protection from T1D development irrespective of the applied dose. In contrast, application of subimmunogenic doses of the strong-agonistic insulin variant in NOD mice with moderate levels of IAA prevented T1D. In NOD mice with high IAA indices at a young age of 4 weeks, Treg generation using the insulin variant was not able to achieve complete prevention of disease development. These data support the view that NOD mice at an age of 4 weeks and with very high indices of IAA show insulin-specific T cell activation, which consequently impairs efficient Treg conversion [97].
Concluding remarks
In autoimmunity such as T1D, it became evident that efficient induction of insulin-specific Foxp3+ Treg cells can be accomplished when strong-agonistic variants of insulin B chain epitopes are used under subimmunogenic conditions. The development of humanized mice models for the careful study of immunotherapeutic approaches in vivo will facilitate our understanding of these approaches in the presence of a human immune system. Based on the mechanistic insight that high doses of antigens promote cell proliferation and activation, it appears reasonable to develop approaches which are able to inhibit T cell activation. These approaches can also be combined with antigen-specific interventions for long-term tolerance induction. Such efforts could result in combinatorial strategy able to limit factors that interfere with the efficient generation of Foxp3+ Treg cells, while maintaining antigen-specificity.
An overview on general approaches for the envisioned targeting of Foxp3+ Treg cells is provided in Figure 2. Future studies will continue to improve our understanding of the involved molecular mechanisms of tolerance induction, with the goal to increase specificity and efficacy of therapies. This will be necessary to meet the complexity of the human immune system.
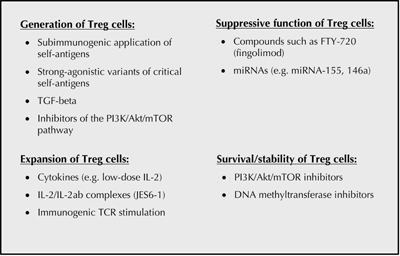 |
|
Figure 2. Approaches for the targeting of regulatory Foxp3+ CD4+ (Treg) cells. Studies in various model systems have helped to develop strategies suitable for the efficient manipulation of Foxp3+ Treg cells. To achieve efficient Foxp3+ Treg cell generation subimmunogenic application of antigens, e.g. strong-agonistic variants of self-antigens, is required. Enhancement of Treg induction can be achieved using transforming growth factor beta (TGF-beta), inhibitors of the phosphoinositide 3-kinase (PI3K)-Akt-mTOR pathways [98], or microRNAs [127]. Low doses of interleukin 2 (IL-2) [128, 129], specific IL-2/IL-2 antibody complexes, and immunogenic stimulation with antigen were found to help the expansion of Treg cells [98, 130]. Strategies for an increase of Treg cell suppressive function might be supported by the use of compounds such as fingolimod (FTY720) [131-134]. Additionally, an improvement in Treg cell survival and stability can be supported by PI3K-Akt/mTOR-inhibitors or by the use of DNA methyltransferase inhibitors [98]. |
|
Disclosure: The authors report no conflict of interests.
Acknowledgments:
BW was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG), grant no. WE 4656/1-1. CD was supported by a Leopoldina research fellowship (BMBF-LPD 9901/8 184), and is now supported by a Junior Research Group at Helmholtz Zentrum München, Germany.
References
- Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010. 464(7293):1293-1300. [DOD] [CrossRef]
- Maahs DM, Rewers M. Mortality and renal disease in type 1 diabetes mellitus - progress made, more to be done. J Clin Endocrinol Metab 2006. 91(10):3757-3759. [DOD] [CrossRef]
- Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care 2003. 26(3):832-836. [DOD] [CrossRef]
- Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 2008. 57(4):1084-1092. [DOD] [CrossRef]
- Concannon P, Chen WM, Julier C, Morahan G, Akolkar B, Erlich HA, Hilner JE, Nerup J, Nierras C, Pociot F, et al. Genome-wide scan for linkage to type 1 diabetes in 2,496 multiplex families from the Type 1 Diabetes Genetics Consortium. Diabetes 2009. 58(4):1018-1022. [DOD] [CrossRef]
- Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity 2010. 32(4):468-478. [DOD] [CrossRef]
- Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. New Engl J Med 2008. 359(26):2849-2850. [DOD] [CrossRef]
- Barker JM, Barriga KJ, Yu L, Miao D, Erlich HA, Norris JM, Eisenbarth GS, Rewers M. Prediction of autoantibody positivity and progression to type 1 diabetes: Diabetes Autoimmunity Study in the Young (DAISY). J Clin Endocrinol Metab 2004. 89(8):3896-3902. [DOD] [CrossRef]
- Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol 2010. 11(1):21-27. [DOD] [CrossRef]
- Sherr J, Sosenko J, Skyler JS, Herold KC. Prevention of type 1 diabetes: the time has come. Nat Clin Pract Endocrinol Metab 2008. 4(6):334-343. [DOD]
- Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996. 45(7):926-933. [DOD] [CrossRef]
- Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, Leslie RD. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001. 44(3):354-362. [DOD] [CrossRef]
- Pugliese A, Zeller M, Fernandez A Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 1997. 15(3):293-297. [DOD] [CrossRef]
- Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, Wickramasinghe S, Colle E, Polychronakos C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet 1997. 15(3):289-292. [DOD] [CrossRef]
- Castano L, Ziegler AG, Ziegler R, Shoelson S, Eisenbarth GS. Characterization of insulin autoantibodies in relatives of patients with type I diabetes. Diabetes 1993. 42(8):1202-1209. [DOD] [CrossRef]
- Alleva DG, Crowe PD, Jin L, Kwok WW, Ling N, Gottschalk M, Conlon PJ, Gottlieb PA, Putnam AL, Gaur A. A disease-associated cellular immune response in type 1 diabetics to an immunodominant epitope of insulin. J Clin Invest 2001. 107(2):173-180. [DOD] [CrossRef]
- Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol 1995. 25(4):1056-1062. [DOD] [CrossRef]
- Wegmann DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol 1994. 24(8):1853-1857. [DOD] [CrossRef]
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005. 435(7039):220-223. [DOD] [CrossRef]
- Moriyama H, Abiru N, Paronen J, Sikora K, Liu E, Miao D, Devendra D, Beilke J, Gianani R, Gill RG, Eisenbarth GS. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci USA 2003. 100(18):10376-10381. [DOD] [CrossRef]
- Martin-Pagola A, Pileggi A, Zahr E, Vendrame F, Damaris MR, Snowhite I, Ricordi C, Eisenbarth GS, Nakayama M, Pugliese A. Insulin2 gene (Ins2) transcription by NOD bone marrow-derived cells does not influence autoimmune diabetes development in NOD-Ins2 knockout mice. Scand J Immunol 2009. 70(5):439-446. [DOD] [CrossRef]
- Thebault-Baumont K, Dubois-Laforgue D, Krief P, Briand JP, Halbout P, Vallon-Geoffroy K, Morin J, Laloux V, Lehuen A, Carel JC, et al. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest 2003. 111(6):851-857. [DOD]
- Kobayashi M, Jasinski J, Liu E, Li M, Miao D, Zhang L, Yu L, Nakayama M, Eisenbarth GS. Conserved T cell receptor alpha-chain induces insulin autoantibodies. Proc Natl Acad Sci USA 2008. 105(29):10090-10094. [DOD] [CrossRef]
- Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, Cuthbertson D, Rafkin-Mervis LE, Chase HP, Leschek E. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial - Type 1. Diabetes Care 2005. 28(5):1068-1076. [DOD] [CrossRef]
- Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 2002. 346(22):1685-1691. [DOD] [CrossRef]
- Harrison LC, Honeyman MC, Steele CE, Stone NL, Sarugeri E, Bonifacio E, Couper JJ, Colman PG. Pancreatic beta-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for type 1 diabetes. Diabetes Care 2004. 27(10):2348-2355. [DOD] [CrossRef]
- Nanto-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, Korhonen S, Erkkola R, Sipilä JI, Haavisto L, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet 2008. 372(9651):1746-1755. [DOD] [CrossRef]
- Lucas JL, Mirshahpanah P, Haas-Stapleton E, Asadullah K, Zollner TM, Numerof RP. Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell Immunol 2009. 257(1-2):97-104. [DOD] [CrossRef]
- Luo X, Herold KC, Miller SD. Immunotherapy of type 1 diabetes: where are we and where should we be going? Immunity 2010. 32(4):488-499. [DOD]
- Bonifacio E, Achenbach P, Pan L, Ziegler AG. Mucosal insulin vaccination for type 1 diabetes prevention. Exp Clin Endocrinol Diabetes 2008. 116(Suppl 1):S26-S29. [DOD] [CrossRef]
- Sigal NH, Dumont FJ. Cyclosporin A, FK-506, and rapamycin: pharmacologic probes of lymphocyte signal transduction. Ann Rev Immunol 1992. 10:519-560. [DOD] [CrossRef]
- Stiller CR, Dupre J, Gent M, Jenner MR, Keown PA, Laupacis A, Martell R, Rodger NW, von Graffenried B, Wolfe BM. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science 1984. 223(4643):1362-1367. [DOD] [CrossRef]
- Assan R, Feutren G, Debray-Sachs M, Quiniou-Debrie MC, Laborie C, Thomas G, Chatenoud L, Bach JF. Metabolic and immunological effects of cyclosporin in recently diagnosed type 1 diabetes mellitus. Lancet 1985. 1(8420):67-71. [DOD] [CrossRef]
- Feutren G, Papoz L, Assan R, Vialettes B, Karsenty G, Vexiau P, Du Rostu H, Rodier M, Sirmai J, Lallemand A, et al Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet 1986. 2(8499):119-124. [DOD] [CrossRef]
- Cyclosporin-induced remission of IDDM after early intervention. Association of 1 yr of cyclosporin treatment with enhanced insulin secretion. The Canadian-European Randomized Control Trial Group. Diabetes 1988. 37(11):1574-1582. [DOD] [CrossRef]
- Martin S, Schernthaner G, Nerup J, Gries FA, Koivisto VA, Dupre J, Standl E, Hamet P, McArthur R, Tan MH, et al. Follow-up of cyclosporin A treatment in type 1 (insulin-dependent) diabetes mellitus: lack of long-term effects. Diabetologia 1991. 34(6):429-434. [DOD] [CrossRef]
- Brazelton TR, Morris RE. Molecular mechanisms of action of new xenobiotic immunosuppressive drugs: tacrolimus (FK506), sirolimus (rapamycin), mycophenolate mofetil and leflunomide. Curr Opin Immunol 1996. 8(5):710-720. [DOD] [CrossRef]
- Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000. 47(2-3):85-118. [DOD] [CrossRef]
- Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, Lang P, Grinyo J, Halloran PF, Solez K, et al. Costimulation blockade with belatacept in renal transplantation. New Engl J Med 2005. 353(8):770-781. [DOD] [CrossRef]
- Gottlieb PA, Quinlan S, Krause-Steinrauf H, Greenbaum CJ, Wilson DM, Rodriguez H, Schatz DA, Moran AM, Lachin JM, Skyler JS. Failure to preserve beta-cell function with mycophenolate mofetil and daclizumab combined therapy in patients with new-onset type 1 diabetes. Diabetes Care 2010. 33(4):826-832. [DOD] [CrossRef]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000. 12(4):431-440. [DOD] [CrossRef]
- Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev 2006. 212:8-27. [DOD] [CrossRef]
- Molina A. A decade of rituximab: improving survival outcomes in non-Hodgkin's lymphoma. Ann Rev Med 2008. 59:237-250. [DOD] [CrossRef]
- Looney RJ. B cells as a therapeutic target in autoimmune diseases other than rheumatoid arthritis. Rheumatology 2005. 44(Suppl 2):ii13-ii17. [DOD] [CrossRef]
- Kazkaz H, Isenberg D. Anti B cell therapy (rituximab) in the treatment of autoimmune diseases. Curr Opin Pharmacol 2004. 4(4):398-402. [DOD] [CrossRef]
- Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. New Engl J Med 2009. 361(22):2143-2152. [DOD] [CrossRef]
- Linsley PS, Nadler SG. The clinical utility of inhibiting CD28-mediated costimulation. Immunol Rev 2009. 229(1):307-321. [DOD] [CrossRef]
- Bluestone JA, St Clair EW, Turka LA. CTLA4Ig: bridging the basic immunology with clinical application. Immunity 2006. 24(3):233-238. [DOD] [CrossRef]
- Herold KC, Vezys V, Koons A, Lenschow D, Thompson C, Bluestone JA. CD28/B7 costimulation regulates autoimmune diabetes induced with multiple low doses of streptozotocin. J Immunol 1997. 158(2):984-991. [DOD]
- Herold KC, Lu J, Rulifson I, Vezys V, Taub D, Grusby MJ, Bluestone JA. Regulation of C-C chemokine production by murine T cells by CD28/B7 costimulation. J Immunol 1997. 159(9):4150-4153. [DOD]
- Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin HY, Fuchs E, Singh B, Thompson CB, Bluestone JA. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity 1996. 5(3):285-293. [DOD] [CrossRef]
- Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, Menter A, Lowe NJ, Krueger G, Brown MJ, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest 1999. 103(9):1243-1252. [DOD] [CrossRef]
- Kremer JM, Genant HK, Moreland LW, Russell AS, Emery P, Abud-Mendoza C, Szechinski J, Li T, Ge Z, Becker JC, Westhovens R. Effects of abatacept in patients with methotrexate-resistant active rheumatoid arthritis: a randomized trial. Ann Intern Med 2006. 144(12):865-876. [DOD]
- Kremer JM, Dougados M, Emery P, Durez P, Sibilia J, Shergy W, Steinfeld S, Tindall E, Becker JC, Li T, et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum 2005. 52(8):2263-2271. [DOD] [CrossRef]
- Kremer JM, Genant HK, Moreland LW, Russell AS, Emery P, Abud-Mendoza C, Szechinski J, Li T, Teng J, Becker JC, Westhovens R. Results of a two-year followup study of patients with rheumatoid arthritis who received a combination of abatacept and methotrexate. Arthritis Rheum 2008. 58(4):953-963. [DOD] [CrossRef]
- Russell AS, Wallenstein GV, Li T, Martin MC, Maclean R, Blaisdell B, Gajria K, Cole JC, Becker JC, Emery P. Abatacept improves both the physical and mental health of patients with rheumatoid arthritis who have inadequate response to methotrexate treatment. Ann Rheum Dis 2007. 66(2):189-194. [DOD] [CrossRef]
- Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011. 378(9789):412-419. [DOD] [CrossRef]
- Kodama S, Davis M, Faustman DL. The therapeutic potential of tumor necrosis factor for autoimmune disease: a mechanistically based hypothesis. Cell Mol Life Sci 2005. 62(16):1850-1862. [DOD] [CrossRef]
- Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, McDevitt HO. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med 1994. 180(3):995-1004. [DOD] [CrossRef]
- Feldmann M, Williams RO, Paleolog E. What have we learnt from targeted anti-TNF therapy? Ann Rheum Dis 2010. 69(Suppl 1):i97- i99. [DOD]
- Tack CJ, Kleijwegt FS, Van Riel PL, Roep BO. Development of type 1 diabetes in a patient treated with anti-TNF-alpha therapy for active rheumatoid arthritis. Diabetologia 2009. 52(7):1442-1444. [DOD] [CrossRef]
- Bloom BJ. Development of diabetes mellitus during etanercept therapy in a child with systemic-onset juvenile rheumatoid arthritis. Arthrit Rheum 2000. 43(11):2606-2608. [DOD] [CrossRef]
- Mastrandrea L, Yu J, Behrens T, Buchlis J, Albini C, Fourtner S, Quattrin T. Etanercept treatment in children with new-onset type 1 diabetes: pilot randomized, placebo-controlled, double-blind study. Diabetes Care 2009. 32(7):1244-1249. [DOD] [CrossRef]
- Nuki G, Bresnihan B, Bear MB, McCabe D. Long-term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthrit Rheum 2002. 46(11):2838-2846. [DOD] [CrossRef]
- Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. New Engl J Med 2007. 356(15):1517-1526. [DOD] [CrossRef]
- Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care 2009. 32(9):1663-1668. [DOD] [CrossRef]
- Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, Kay TW. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes 2004. 53(1):113-121. [DOD] [CrossRef]
- Mandrup-Poulsen T, Pickersgill L, Donath MY. Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol 2010. 6(3):158-166. [DOD] [CrossRef]
- Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol 2007. 7(8):622-632. [DOD] [CrossRef]
- Cosimi AB, Burton RC, Colvin RB, Goldstein G, Delmonico FL, LaQuaglia MP, Tolkoff-Rubin N, Rubin RH, Herrin JT, Russell PS. Treatment of acute renal allograft rejection with OKT3 monoclonal antibody. Transplantation 1981. 32(6):535-539. [DOD] [CrossRef]
- Friend PJ, Hale G, Chatenoud L, Rebello P, Bradley J, Thiru S, Phillips JM, Waldmann H. Phase I study of an engineered aglycosylated humanized CD3 antibody in renal transplant rejection. Transplantation 1999. 68(11):1632-1637. [DOD] [CrossRef]
- Abramowicz D, Schandene L, Goldman M, Crusiaux A, Vereerstraeten P, De Pauw L, Wybran J, Kinnaert P, Dupont E, Toussaint C. Release of tumor necrosis factor, interleukin-2, and gamma-interferon in serum after injection of OKT3 monoclonal antibody in kidney transplant recipients. Transplantation 1989. 47(4):606-608. [DOD] [CrossRef]
- Alegre ML, Peterson LJ, Xu D, Sattar HA, Jeyarajah DR, Kowalkowski K, Thistlethwaite JR, Zivin RA, Jolliffe L, Bluestone JA. A non-activating "humanized" anti-CD3 monoclonal antibody retains immunosuppressive properties in vivo. Transplantation 1994. 57(11):1537-1543. [DOD]
- Bolt S, Routledge E, Lloyd I, Chatenoud L, Pope H, Gorman SD, Clark M, Waldmann H. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol 1993. 23(2):403-411. [DOD] [CrossRef]
- Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002. 346(22):1692-1698. [DOD] [CrossRef]
- Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005. 352(25):2598-2608. [DOD] [CrossRef]
- Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005. 54(6):1763-1769. [DOD] [CrossRef]
- Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr, Bode B, Aronoff S, Holland C, Carlin D, et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011. 378(9790):487-497. [DOD] [CrossRef]
- Burnet FM. The clonal selection theory. Cambridge Press, 1959. [DOD]
- Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell 1987. 49(2):273-280. [DOD] [CrossRef]
- Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature 1988. 333(6175):742-746. [DOD] [CrossRef]
- Lederberg J. Genes and antibodies. Science 1959. 129(3364):1649-1653. [DOD] [CrossRef]
- Ohki H, Martin C, Corbel C, Coltey M, Le Douarin NM. Tolerance induced by thymic epithelial grafts in birds. Science 1987. 237(4818):1032-1035. [DOD] [CrossRef]
- Salaun J, Bandeira A, Khazaal I, Calman F, Coltey M, Coutinho A, Le Douarin NM. Thymic epithelium tolerizes for histocompatibility antigens. Science 1990. 247(4949 Pt 1):1471-1474. [DOD] [CrossRef]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995. 155(3):1151-1164. [DOD]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003. 4(4):330-336. [DOD] [CrossRef]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003. 299(5609):1057-1061. [DOD] [CrossRef]
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 2003. 4(4):337-342. [DOD] [CrossRef]
- Ochs HD, Ziegler SF, Torgerson TR. FOXP3 acts as a rheostat of the immune response. Immunol Rev 2005. 203:156-164. [DOD] [CrossRef]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001. 27(1):68-73. [DOD] [CrossRef]
- Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 2007. 204(1):57-63. [DOD] [CrossRef]
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 2007. 8(2):191-197. [DOD] [CrossRef]
- Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol 2002. 3(8):756-763. [DOD]
- Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol 2001. 2(4):301-306. [DOD] [CrossRef]
- Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med 2004. 199(10):1401-1408. [DOD] [CrossRef]
- Daniel C, von Boehmer H. Extrathymic regulatory T cells - chances and challenges for prevention of autoimmune disease. Adv Immunol 2011. 112:177-213. [DOD] [CrossRef]
- Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med 2011. 208(7):1501-1510. [DOD] [CrossRef]
- Daniel C, Wennhold K, Kim HJ, von Boehmer H. Enhancement of antigen-specific Treg vaccination in vivo. Proc Natl Acad Sci USA 2010. 107(37):16246-16251. [DOD] [CrossRef]
- Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol 2005. 6(12):1219-1227. [DOD] [CrossRef]
- Verginis P, McLaughlin KA, Wucherpfennig KW, von Boehmer H, Apostolou I. Induction of antigen-specific regulatory T cells in wild-type mice: visualization and targets of suppression. Proc Natl Acad Sci USA 2008. 105(9):3479-3484. [DOD] [CrossRef]
- Merkenschlager M, von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med 2010. 207(7):1347-1350. [DOD] [CrossRef]
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O'Connor E, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA 2008. 105(22):7797-7802. [DOD] [CrossRef]
- Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, Fairchild PJ, Mellor AL, Ron D, Waldmann H. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci USA 2009. 106(29):12055-12060. [DOD] [CrossRef]
- Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 2008. 205(3):565-574. [DOD] [CrossRef]
- Klein L, Khazaie K, von Behmer H. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc Natl Acad Sci USA 2003. 100(15):8886-8891. [DOD] [CrossRef]
- Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol 2008. 38(6):1654-1663. [DOD] [CrossRef]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science 2010. 329(5999):1667-1671. [DOD] [CrossRef]
- Fairchild PJ, Wildgoose R, Atherton E, Webb S, Wraith DC. An autoantigenic T cell epitope forms unstable complexes with class II MHC: a novel route for escape from tolerance induction. Int Immunol 1993. 5(9):1151-1158. [DOD] [CrossRef]
- Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol 1999. 17:369-397. [DOD] [CrossRef]
- Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol 2005. 6(5):490-496. [DOD] [CrossRef]
- Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity 1995. 3(4):407-415. [DOD] [CrossRef]
- Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010. 11(3):225-231. [DOD] [CrossRef]
- Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci USA 2010. 107(24):10978-10983. [DOD] [CrossRef]
- Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, Marrack P, Eisenbarth G, Kappler JW. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci USA 2011. 108(40):16729-16734. [DOD] [CrossRef]
- He XL, Radu C, Sidney J, Sette A, Ward ES, Garcia KC. Structural snapshot of aberrant antigen presentation linked to autoimmunity: the immunodominant epitope of MBP complexed with I-Au. Immunity 2002. 17(1):83-94. [DOD] [CrossRef]
- Sethi DK, Schubert DA, Anders AK, Heroux A, Bonsor DA, Thomas CP, Sundberg EJ, Pyrdol J, Wucherpfennig KW. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med 2011. 208(1):91-102. [DOD] [CrossRef]
- Wucherpfennig KW, Sethi D. T cell receptor recognition of self and foreign antigens in the induction of autoimmunity. Semin Immunol 2011. 23(2):84-91. [DOD] [CrossRef]
- Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol 2010. 11(4):350-354. [DOD] [CrossRef]
- Jaeckel E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat Immunol 2004. 5(10):1028-1035. [DOD] [CrossRef]
- Abiru N, Wegmann D, Kawasaki E, Gottlieb P, Simone E, Eisenbarth GS. Dual overlapping peptides recognized by insulin peptide B:9-23 T cell receptor AV13S3 T cell clones of the NOD mouse. J Autoimmun 2000. 14(3):231-237. [DOD] [CrossRef]
- Burton AR, Vincent E, Arnold PY, Lennon GP, Smeltzer M, Li CS, Haskins K, Hutton J, Tisch RM, Sercarz EE, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes 2008. 57(5):1321-1330. [DOD] [CrossRef]
- Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol 2007. 178(10):6051-6057. [DOD]
- Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, Wilson IA, Teyton L. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science 2000. 288(5465):505-511. [DOD] [CrossRef]
- Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, Fremont DH. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity 2000. 12(6):699-710. [DOD] [CrossRef]
- Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol 2001. 2(6):501-507. [DOD] [CrossRef]
- Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med 2010. 207(8):1701-1711. [DOD] [CrossRef]
- von Boehmer H, Daniel C. Therapeutic opportunities for manipulating T(Reg) cells in autoimmunity and cancer. Nat Rev Drug Discov 2012. 12(1):51-63. [DOD] [CrossRef]
- Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP 3rd, Armand P, Cutler C, Ho VT, Treister NS. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med 2011. 365(22):2055-2066. [DOD] [CrossRef]
- Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, Sene D, Cacoub P, Klatzmann D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 2011. 365(22):2067-2077. [DOD] [CrossRef]
- Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, Sprent J. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med 2009. 206(4):751-760. [DOD] [CrossRef]
- Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol 2010. 11(11):1047-1056. [DOD] [CrossRef]
- Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, Chi H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol 2009. 10(7):769-777. [DOD] [CrossRef]
- Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol 2007. 178(4):2458-2468. [DOD]
- Sawicka E, Dubois G, Jarai G, Edwards M, Thomas M, Nicholls A, Albert R, Newson C, Brinkmann V, Walker C. The sphingosine 1-phosphate receptor agonist FTY720 differentially affects the sequestration of CD4+/CD25+ T-regulatory cells and enhances their functional activity. J Immunol 2005. 175(12):7973-7980. [DOD]
This article has been cited by other articles:

|
The effect of growth hormone treatment on islet mass in streptozotocin treated mice
Scheinman EJ, Damouni R, Caspi A, Shen-Orr Z, Tiosano D, LeRoith D
Diabetes Metab Res Rev 2014. In press
|
|

|
Alternative Medicine in Diabetes - Role of Angiogenesis, Oxidative Stress, and Chronic Inflammation
El-Refaei MF, Abduljawad SH, Alghamdi AH
Rev Diabet Stud 2014. 11(3-4):231-244
|
|

|
Targeted Antigen Delivery to DEC-205(+) Dendritic Cells for Tolerogenic Vaccination
Petzold C, Schallenberg S, Stern JN, Kretschmer K
Rev Diabet Stud 2012. 9(4):305-318
|
|
|


)
)

