Review
| Rev Diabet Stud,
2007,
4(2):77-84 |
DOI 10.1900/RDS.2007.4.77 |
Congenital and Environmental Factors Associated with Adipocyte Dysregulation as Defects of Insulin Resistance
Chao-Ping Wang1, Fu-Mei Chung1, Shyi-Jang Shin2, Yau-Jiunn Lee3
1Division of Cardiology, Department of Internal Medicine, E-Da Hospital, I-Shou University, Kaohsiung, 82445 Taiwan.
2Department of Internal Medicine, Kaohsiung Medical University, Kaohsiung, 80307 Taiwan.
3Lee´s Endocrinologic Clinic, Pingtung, 90000 Taiwan.
Address correspondence to: Yau-Jiunn Lee, e-mail: t3275@ms25.hinet.net
Keywords: diabetes, insulin resistance, metabolic syndrome, adipocyte dysregulation
Abstract
The metabolic syndrome refers to insulin resistance and its associated cluster of related cardiovascular metabolic risk factors including type 2 diabetes, hypertension, dyslipidemia and central obesity. Although many hypotheses and facts have been proposed to explain the interaction between genetic and environmental causes of this syndrome, the primary etiology of the metabolic syndrome is adipose tissue dysregulation. Firstly, the thrifty genotype and phenotype hypothesis may explain the endemic increase in type 2 diabetes and cardiovascular disease in developing countries and may elucidate congenital susceptibility to and environmental triggering of the metabolic syndrome. Secondly, overnutrition leads to fatty acid (FA) accumulation in adipocytes and to an overflow to ectopic fat storage organs. This causes functional changes in adipocytes shifting the intra-cellular metabolic pathway toward insulin resistance. Thirdly, obese subjects exhibit increased fat cell size and over-secretion of biologic adipocytokines. Fourthly, failure to adequately develop adipose tissue mass, as seen in lipodystrophy cases, causes severe insulin resistance and diabetes. Lastly, similarly to human type 2 diabetes, Psammonys obesus, a desert rat which feeds mainly on low-calorie vegetation, develops the metabolic syndrome when given a diet of calorie-rich food. The above evidence indicates that adipocyte dysregulation and secretion of FA as well as certain molecules from overloaded adipocytes-adipokines contribute to the pathogenesis of impaired insulin secretion and insulin resistance, endothelial dysfunction, a pro-inflammatory state and promote progression of atherosclerosis. The metabolic syndrome is a modern disease resulting in adipocyte dysmetabolism which originates from the paradox of slow human evolution combined with rapid environmental changes.
Introduction
The metabolic syndrome refers to the cluster of several cardio-metabolic risk factors, including central obesity, hyperglycemia, dyslipidemia (elevated triglycerides, low levels of high density lipoprotein cholesterol) and elevated blood pressure, all of which have been linked to insulin resistance [1]. The metabolic syndrome is a common metabolic disorder resulting from the increased prevalence of obesity. The disorder is defined in various ways and different definitions require different cut points and have different mandatory inclusion criteria [2]. The apparent pathophysiology is insulin resistance with excessive flux of fatty acids. Insulin resistance was brought to the fore by Gerald Reaven in 1988 [3]. Reaven referred to the disorder as Syndrome X, but it is also commonly known as Reaven’s syndrome, insulin resistance syndrome and metabolic syndrome. The latter is the most widely used terminology.
The first operational definition of the metabolic syndrome proposed by the World Health Organization [4] was hyperglycemia and/or insulin resistance as a central feature associated with two or more related metabolic abnormalities. The metabolic syndrome has also been defined by readily assessable clinical and biochemical measures to improve identification of at-risk subjects [5, 6]. Large epidemiological surveys show that the metabolic syndrome, according to most definitions, is common [7]. The prevalence of the metabolic syndrome increases with age and varies with gender and ethnicity [8]. Prospective studies have established that the metabolic syndrome is associated with a two-fold increased risk in cardiovascular disease [9]. Proposed pathophysiological explanations for the metabolic syndrome include insulin resistance, chronic inflammation and ectopic fat accumulation following adipose tissue saturation with fatty acids [10]. However, the central core of the metabolic syndrome may be failed evolutionary adaptation of adipose tissue to affluent lifestyles and excess energy intake.
Adipose tissue in human evolution
Adipose tissue is traditionally viewed as a site of excess energy stored as triglycerides (TGs). When needed elsewhere in the body, that energy is released as fatty acids (FAs) [11-13]. Although often cited in human evolutionary theory, the role of the adipose tissue in human evolution has not been clearly elucidated. As adipose tissue is not fossilized, clinical studies have relied on comparative data [13]. Nowadays, fat depots of normal persons contain sufficient energy to survive one month or more of total starvation. This excess energy storage is less important now since food supplies are plentiful. In previous eras of human history, long periods of famine after years of bad harvests and long winters claimed many lives [14]. Those who survived despite limited external energy supplies were those with large fat deposits. Genetic selection favored those who could accumulate sufficient excess fat to survive long periods of starvation [15]. Such conditions may have caused selection of a so called "thrifty gene", which, in the present era of abundant food supplies, exacerbates obesity [16, 17]. This selection of obesity genes by evolution (congenital factor) and overnutrition due to an affluent life style (environmental factor) may be the leading causes of severe obesity worldwide and particularly in third world countries [18]. The metabolic syndrome was thus introduced into modern civilization (Figure 1).
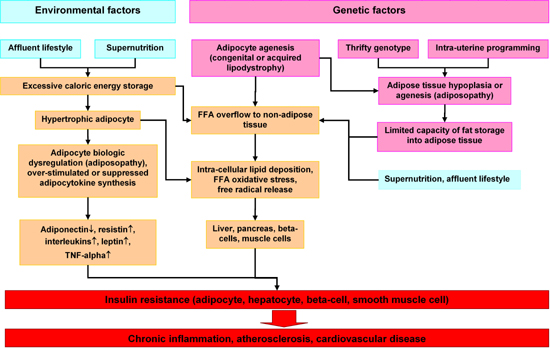 |
 |
Figure 1. Hypothetical program of adipocyte dysregulation (adiposopathy) in the pathogenesis of the insulin resistance syndrome. Environmental and genetic factors may work in concert to cause dysregulation of hypertrophic adipocytes, FFA overflow in non-adipose tissue such as liver and pancreas and finally insulin resistance and chronic diseases. |
|
Thrifty genotype and phenotype hypothesis
A rapidly growing epidemic of type 2 diabetes and cardiovascular disease has afflicted developing countries throughout Southeast Asia [19]. By 2025, India is predicted to have >60 million diabetic patients, and coronary heart disease (CHD) is expected to be the leading cause of adult mortality [20, 21]. In other words, one in five diabetic patients worldwide are expected be Indian, and three in four from developing countries [21]. This phenomenal rise in diabetes and CHD has been ascribed to rapid demographic, nutritional and socioeconomic changes, the so-called epidemiological, nutritional and economic transition [22].
The thrifty genotype and the thrifty phenotype are two nonexclusive explanations for the insulin resistance epidemic in developing countries [16, 23-25]. Asian populations have faced undernutrition for many generations; however, the diabetes epidemic is of recent origin [19]. Susceptibility to diabetes is ascribed to the evolution of thrifty genes, which enhanced survival in the past when food supplies were scarce and intermittent but which have become physically detrimental in contemporary conditions of plentiful food and sedentary lifestyles [23-26].
The evolutionary advantage of Homo sapiens lay in the way he obtained food, which in turn depended on physical activity. A consistent food supply, however, could never be relied upon. Certain genes evolved to regulate the efficient intake and utilization of fuel stores, thus ensuring survival during famine. These genes were called "thrifty genes" in 1962 [16]. In addition there is evidence that the human genome has remained essentially unchanged for as long as 10,000 years and certainly over the past 40-100 years [26, 27]. Although the absolute caloric intake of modern humans is probably lower than that of early hunter-gatherers, the caloric balance in modern adult populations is nevertheless positive because of the sedentary lifestyle in the today’s society [17, 26]. The combination of an abundant and reliable food supply and lack of physical activity eliminates the biochemical cycles which evolved from feast-famine and physical activity-rest cycles. Hence, certain metabolic processes no longer occur in cycles, which ultimately lead to metabolic disorders such as obesity and type 2 diabetes [17, 26].
An alternative explanation is the recently proposed thrifty phenotype hypothesis which ascribes the obesity epidemic to an unfavorable intrauterine environment [23, 24, 27]. The crucial importance of the intrauterine period in determining health and disease is understandable given that humans begin life as a single cell and that more than three quarters of total cell divisions occur in utero [28, 29]. Substantial growth and development is completed before birth, as demonstrated by the inverse relationship between birth weight and risk of diabetes and metabolic syndrome later in life [30-32]. The thrifty phenotype hypothesis proposes that early-life metabolic adaptations promote survival, with the developing organism responding to environmental cues by selecting an appropriate growth trajectory [33, 34].
The two explanations are not necessarily mutually exclusive and may be complimentary. It has been said that the rapidly emerging epidemic of the metabolic syndrome in the twentieth Century reflects the rapidly accelerating pace of niche construction relative to the slower physiological combination of developmental plasticity and parental effects [17].
Environmental factors - overnutrition, obesity and adipocyte hypertrophy
Adipose tissue becomes important when increased energy storage is required. However, these deposits are fairly limited. Although the excess energy may be efficiently stored, fat storage capacity is limited [35, 36]. The varying functional capacity of adipose tissue explains the incomplete overlapping between the metabolic syndrome and obesity. Obesity is characterized by elevated fat accumulation caused by enlarged adipocytes [37-39]. The typical adipocyte without accumulated fat is approximately 10-12 micrometers in diameter, the approximate size of a lymphocyte. After normal fat accumulation, the diameter increases 10-fold, a 1000-fold volume increase [39].
Researchers have demonstrated associations between adipocyte enlargement and metabolic change, e.g. lipid metabolism, as well as the release of cytokines, chemokines and many other biologically active molecules, commonly known as adipokines [40, 41]. These secreted products may participate in developing a chronic low-grade inflammatory state which is "common soil" for the pathogenesis of metabolic and cardiovascular complications of obesity [35, 42-46].
The effects of ectopic fat deposition and the overflow of lipids into muscles and liver on the function of beta-cells
For the first time in history, large human populations have a readily available, inexpensive and palatable food supply. This has created a situation, on a scale not previously encountered, in which the capacity to store triglycerides in adipocytes has become an important determinant of human health. An inadequate number of adipocytes (e.g., lipodystrophia) or overcapacity of adipocytes (e.g., severe obesity), produce similar and unfavorable metabolic effects where ectopic triglyceride stores may appear in liver, muscle and pancreatic islet tissue [47, 48].
Clearly, under normal conditions, the storage and release of TG and FA, respectively, are both coordinated and tightly regulated such that lipid fuels are stored during the immediate postprandial periods and released during periods of fasting [49]. More recent studies have demonstrated that impaired regulation of storage and release of energy by adipose tissue elevate plasma FA levels, in particular when the release of FA becomes dissociated from energy requirements in other organs and tissues. Excessive metabolism of FA, including storage of TG, occurs in nonadipose tissues [47, 50]. The expanded view of ectopic metabolism or accumulation of FA or TG is that it causes dysfunction, or lipotoxicity, in these organs and tissues [51-54].
Clinical evidence indicates that overflow to non-adipose tissue results from the limited storage capacity of congenital adipocytes, either from intra-uterine programming or excess energy intake due to an affluent life style and overeating, which can trigger the process of insulin resistance. Firstly, the failure to develop adequate adipose tissue mass (also known as 'lipodystrophy') causes severe insulin resistance and diabetes [55] due to ectopic storage of lipids in the liver, skeletal muscle and pancreatic insulin-secreting beta-cell [56, 57].
Secondly, most obese patients also shunt lipids into skeletal muscle, the liver and apparently into beta-cells [58-60]. The importance of this finding is exemplified by several studies demonstrating that the degree of lipid infiltration into skeletal muscle and the liver highly correlates with insulin resistance [61].
The role of increased fatty acid flux in muscle and insulin resistance
Excess FA trapping in muscles combined with reduced FA oxidation increases TG synthesis. Indeed, increased intramyocellular TG correlates strongly with insulin resistance [52, 54, 61]. Although some studies suggest that intramyocellular TG is a lipotoxic substance in muscle tissue, it more likely functions only as a marker of reduced oxidation relative to FA availability [62]. Of interest in this regard are findings that insulin-resistant and diabetic individuals have a decreased fractional FA uptake and, in some cases, absolute FA uptake by muscles has been observed [52, 63]. However, normal or only slightly reduced plasma FA levels during the immediate postprandial period in insulin-resistant individuals, when plasma FA levels should be dramatically suppressed, combined with modestly reduced FA extraction by the muscle, can still cause excess FA in muscle [64]. Additionally, considerable evidence demonstrates that insulin-resistant individuals exhibit defective mitochondrial oxidation of FA [52, 54], which is reversible by weight loss and exercise [65].
The role of increased fatty acid flux to beta-cells and defects in insulin secretion
Plasma FA can be taken up by pancreatic beta-cells where they can stimulate insulin secretion under certain conditions [66]. The latter apparently occurs because FAs generate long-chain fatty acyl CoA, which contributes to the synthesis of signaling molecules such as DAG and phosphatidic acid or directly stimulates PKC isoforms involved in insulin secretion [67]. Of particular relevance to insulin resistance is the finding that circulating FA are critical in the pancreatic response to glucose in the fasting state [68, 69]. Thus, the high basal insulin response characteristic of insulin resistance might be partly due to increased FA levels before administration. High-plasma FA uptake by beta-cells also increases FA storage as TG [67]. Clinical evidence clearly shows that continued high-cytosolic FA concentrations ultimately decrease insulin secretion [67]. Finally, animal models indicate that chronic exposure of beta-cells to high FA concentrations and glucose causes apoptosis in ZDF rats [51, 66, 70].
Non-alcoholic fatty liver disease - the role of increased fatty acid flux to hepatocytes
Non-alcoholic fatty liver disease (NAFLD) is an ectopic fat storage disorder strongly associated with insulin resistance [71, 72]. Patients with NAFLD are apparently predisposed to type 2 diabetes because of its close association with insulin resistance and its consequences, including hyperinsulinemia, glucose intolerance, hypertriglyceridemia, decreased high-density lipoprotein cholesterol concentration and hypertension [73]. Although most commonly associated with obesity, NAFLD may also be observed in non-obese subjects [74, 75].
Convincing evidence suggests that ectopic fat accumulation in insulin-sensitive tissues is associated with insulin resistance, independent of overall obesity. However, the precise causes and mechanisms underlying fat accumulation in skeletal muscle and the liver are unclear [76]. Identifying why some individuals store fat in insulin-sensitive tissues may be of great importance in developing new insulin-sensitizing agents and for optimizing current therapies [77, 78].
The published literature clearly demonstrates that dysregulated partitioning of FA between storage sites (adipocytes) and sites of utilization (muscle, liver and beta-cells) upsets lipid homeostasis in the latter tissues. Deleterious outcomes include TG accumulation and insulin resistance [47, 48, 57, 58]. This fact suggests that, in the metabolic syndrome, adipose tissue insulin resistance is the only cause of insulin resistance in other sites [79]. Thus, inadequate energy storage in adipocytes causes abnormal lipid metabolism and possibly insulin resistance, inflammation and cell death in alternative storage sites [80].
Lipodystrophy
Lipodystrophies are rare, inherited and acquired disorders characterized by the selective loss of adipose tissue [81]. The main forms can be classified according to their origin, either genetic or acquired, and subclassified by their clinical pattern [36, 55, 81]. Interestingly, the selective loss of adipose tissue is also frequently associated with marked insulin resistance and complications such as type 2 diabetes, dyslipidemia and hypertension [56, 82]. The final outcome of loss of physiological lipid partitioning is total lipodystrophy, a disorder characterized by the absence of adipose tissue [36]. Interestingly, patients with total lipodystrophy, who have no intra-abdominal fat or visceral fat, are severely insulin-resistant. Partial lipodystrophy patients may also exhibit ectopic fat deposition in several organs as well as visceral fat, hypertriglyceridemia and insulin resistance [82].
Despite marked phenotypic and genotypic heterogeneity, most lipodystrophic syndromes are predisposed to metabolic complications commonly observed in obese patients, such as insulin resistance, diabetes mellitus, hepatic steatosis and dyslipidemia [56]. Thus, normal levels of adipose tissue and normal fat distribution are apparently critical for optimizing regulation of lipid and energy metabolism [83]. The precise mechanisms by which the lack of adipose tissue causes hypertriglyceridemia remain unknown. Anecdotal kinetic studies in hyperglycemic patients with lipodystrophies reveal accelerated lipolysis and increased free fatty acid turnover, which drives hepatic triglyceride and very low-density lipoprotein synthesis. Other possible causes of dyslipidemia and ectopic triglyceride accumulation in the liver and skeletal muscles have been proposed but require further confirmation [81].
Adipocytokines
Although adipose tissue was considered inert tissue until 40 years ago, several vital functions have been associated with this highly active tissue [11, 35, 84]. Adipocytokines are bioactive mediators released from adipose tissue including adipocytes and other cells present in fat tissues [85-87]. These include several novel and highly active molecules abundantly released by adipocytes, such as leptin, resistin, adiponectin or visfatin as well as other well known cytokines possibly released by inflammatory cells infiltrating fat, such as TNF-alpha, IL-6, MCP-1 and IL-1 [41, 43]. Leptin and adiponectin are the most abundantly expressed adipocytokines within the adipose tissue [88-90].
These molecules, along with FA, significantly affect total body glucose, lipid metabolism and insulin sensitivity [85, 91, 92]. Adipocytokine release may elucidate the relationship between obesity and cardiovascular phenotypes, including hypertension and atherosclerosis, mainly through their ability to affect and modify endothelial and vascular function as well as by their modulating effects on immune functions, which have been elucidated in several reviews [91-94].
Psammomys obesus, an animal model of environmental-gene interaction of type 2 diabetes
Psammomys obesus, a rat found in semi-desert regions of North African and the Eastern Mediterranean, exhibits insulin resistance similar to that in human type 2 diabetes. In its native habitat, P. obesus feeds mainly on the low-calorie Atriplex halimus plant, and is neither obese nor hyperglycemic. However, when given calorie-rich rodent food feed ad libitum in captivity, it tends to become moderately obese and develops postprandial hyperglycemia [95-97].
The P. obesus model elucidates environmental and genetic factors in the pathogenesis of insulin resistance [95]. P. obesus is characterized by muscle insulin resistance and impaired insulin signaling on a high-energy diet. Insulin resistance imposes a vicious cycle of hyperglycemia and compensatory hyperinsulinemia, leading to beta-cell failure and increased proinsulin secretion [96].
Conclusions
In recent decades, the prevalence of insulin resistance and diabetes has increased in Western populations as well as in Asian populations adopting "Western" lifestyles. This trend indicates the impact of environmental factors such as diet, obesity and physical activity on the pathogenesis of the metabolic syndrome. According to the ‘thrifty gene’ hypothesis, a cluster of genetic defects, which originally provided some populations with a genetic advantage, could predispose some ethnic groups to insulin resistance and diabetes in the presence of an increased food supply. This concept has arisen from hypothesized interactions between genetic, intrauterine and environmental factors.
The inherited defects resulting from adipose tissue differentiation are not fully adapted to modern lifestyles and excess caloric intake. Inadequate adipose tissue mass and overnutrition cause adipocytes to increase fat cell size and stimulate release of insulin-resistant adipocytokines. These processes cause severe insulin resistance, endothelial dysfunction, a proinflammatory state and promote the progression of atherosclerosis. Overnutrition as excess FA overflow to ectopic fat storage organs, such as the liver, muscle, and pancreatic beta-cells, cause functional alternation in these cells and shift the intra-cellular metabolic pathway towards insulin resistance.
Cumulatively, these observations indicate that the 'adipocyte dysregulation-adiposopathy' hypothesis actually identifies congenital or environmental triggering factors as primary causes of insulin resistance.
Acknowledgments:
The authors would like to thank the National Science Council of the Republic of China, Taiwan, for financially supporting this research under contract no. NSC-95-2314-B-475-002-MY3.
References
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005. 365:1415-1428. [DOD] [CrossRef]
- Chew GT, Gan SK, Watts GF. Revisiting the metabolic syndrome. Med J Aust 2006. 185:445-449. [DOD]
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988. 37:1595-1607. [DOD] [CrossRef]
- World Health Organization. Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Part 1: diagnosis and classification of diabetes mellitus. Geneva: WHO, 1999. [DOD]
- Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC Jr, Spertus JA, Costa F. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005. 112(17):2735-2752. [DOD] [CrossRef]
- Alberti KG, Zimmet P, Shaw J. The metabolic syndrome - a new worldwide definition. Lancet 2005. 366(9491):1059-1062. [DOD] [CrossRef]
- Ford ES. Prevalence of the metabolic syndrome defined by the International Diabetes Federation among adults in the US. Diabetes Care 2005. 28:2745-2749. [DOD] [CrossRef]
- Kolovou GD, Anagnostopoulou KK, Salpea KD, Mikhailidis DP. The prevalence of metabolic syndrome in various populations. Am J Med Sci 2007. 333(6):362-371. [DOD] [CrossRef]
- Gami AS, Witt BJ, Howard DE, Erwin PJ, Gami LA, Somers VK, Montori VM. Metabolic syndrome and risk of incident cardiovascular events and death: a systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol 2007. 49:403-414. [DOD] [CrossRef]
- Laclaustra M, Corella D, Ordovas JM. Metabolic syndrome pathophysiology: the role of adipose tissue. Nutr Metab Cardiovasc Dis 2007. 17:125-139. [DOD] [CrossRef]
- Fruhbeck G, Gomez-Ambrosi J, Muruzabal FJ, Burrell MA. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am J Physiol Endocrinol Metab 2001. 280:E827-E847. [DOD]
- Badman MK, Flier JS. The adipocyte as an active participant in energy balance and metabolism. Gastroenterology 2007. 132:2103-2115. [DOD] [CrossRef]
- Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002. 45:1201-1210. [DOD] [CrossRef]
- Unger RH. Longevity, lipotoxicity and leptin: the adipocyte defense against feasting and famine. Biochimie 2005. 87:57-64. [DOD] [CrossRef]
- Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006. 444:847-853. [DOD] [CrossRef]
- Neel JV. Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress"? Am J Hum Genet 1962. 14:353-362. [DOD]
- Chakravarthy MV, Booth FW. Eating, exercise, and "thrifty" genotypes: connecting the dots toward an evolutionary understanding of modern chronic diseases. J Appl Physiol 2004. 96:3-10. [DOD] [CrossRef]
- Kitano H, Oda K, Kimura T, Matsuoka Y, Csete M, Doyle J, Muramatsu M. Metabolic syndrome and robustness tradeoffs. Diabetes 2004. 53:S6-S15. [DOD] [CrossRef]
- Ramachandran A, Snehalatha C, Kapur A, Vijay V, Mohan V, Das AK, Rao PV, Yajnik CS, Prasanna Kuma KM, Nair JD. High prevalence of diabetes and impaired glucose tolerance in India: National Urban Diabetes Survey. Diabetologia 2001. 9:1094-1101. [DOD] [CrossRef]
- Reddy KS. Cardiovascular diseases in India. World Health Stat Q 1993. 46:101-107. [DOD]
- King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates and projections. Diabetes Care 1998. 21(9):1414-1431. [DOD] [CrossRef]
- Yajnik CS. Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. J Nutr 2004. 134:205-210. [DOD]
- Hales CN, Barker DJ. Type 2 (non-insulin dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 1992. 35:595-601. [DOD] [CrossRef]
- Hales CH, Barke DJ. The thrifty phenotype hypothesis. Brit Med Bull 2001. 60:51-67. [DOD] [CrossRef]
- Groop LC, Eriksson JG. The etiology and pathogenesis of non-insulin-dependent diabetes. Ann Med 1992. 24:483-489. [DOD]
- Prentice AM. Early influences on human energy regulation: thrifty genotypes and thrifty phenotypes. Physiol Behav 2005. 86:640-645. [DOD] [CrossRef]
- Barker DJ. Fetal origins of coronary heart disease. Br Med J 1995. 311:171-174. [DOD]
- Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birth weight with diabetes and vascular disease. Lancet 1999. 353(9166):1789-1792. [DOD] [CrossRef]
- Valsamakis G, Knanaka-Gantenbein C, Malamitsi-Puchner A, Mastorakos G. Causes of intrauterine growth restriction and the postnatal development of the metabolic syndrome. Ann N Y Acad Sci 2006. 1092:138-147. [DOD] [CrossRef]
- Fall CHD, Stein CE, Kumaran K, Cox V, Osmond C, Barker DJ. Hales CN. Size at birth, maternal weight, and non-insulin dependent diabetes in South India. Diab Med 1981. 15:220-227. [DOD] [CrossRef]
- Mi J, Law C, Zhang KL, Osmond C, Stein C, Barker D. Effects of infant birth weight and maternal body mass index in pregnancy on components of the insulin resistance syndrome in China. Ann Intern Med 2000. 132:253-260. [DOD]
- Wei JN, Sung FC, Li CY, Chang CH, Lin RS, Lin CC, Chiang CC, Chuang LM. Low birth weight and high birth weight infants are both at an increased risk to have type 2 diabetes among schoolchildren in Taiwan. Diabetes Care 2003. 26:343-348. [DOD] [CrossRef]
- Paradies YC, Montoya MJ, Fullerton SM. Racialized genetics and the study of complex diseases: the thrifty genotype revisited. Perspect Biol Med 2007. 50:203-227. [DOD] [CrossRef]
- Wells JC. The thrifty phenotype as an adaptive maternal effect. Biol Rev Camb Philos Soc 2007. 82:143-172. [DOD]
- Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004. 89:2548-2556. [DOD] [CrossRef]
- Garg A. Adipose tissue dysfunction in obesity and lipodystrophy. Clin Cornerstone 2006. 8:S7-S13. [DOD] [CrossRef]
- Brook CG, Lloyd JK, Wolf WO. Relation between age of onset of obesity and size and number of adipose cells. Br Med J 1972. 2:25-27. [DOD]
- Hirsch J, Batchelor B. Adipose tissue cellularity in human obesity. Clin Endocrinol Metab 1976. 5:299-311. [DOD] [CrossRef]
- Marques BG, Hausman DB, Martin RJ. Association of fat cell size and paracrine growth factors in development of hyperplastic obesity. Am J Physiol 1998. 275:R1898-R1908. [DOD]
- Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab 2007. 92:1023-1033. [DOD] [CrossRef]
- Trayhurn P. Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand 2005. 184:285-293. [DOD] [CrossRef]
- Guerre-Millo M. Adipose tissue and adipokines: for better or worse. Diabetes Metab 2004. 30:13-19. [DOD] [CrossRef]
- Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 2004. 92:347-355. [DOD] [CrossRef]
- Hauner H. Secretory factors from human adipose tissue and their functional role. Proc Nutr Soc 2005. 64:163-169. [DOD] [CrossRef]
- Cederberg A, Enerback S. Insulin resistance and type 2 diabetes - an adipocentric view. Curr Mol Med 2003. 3:107-125. [DOD] [CrossRef]
- Mohammed-Ali V, Pinkney JH, Coppack SW. Adipose tissue as an endocrine and paracrine organ. Int J Obes Rel Metab Disord 1998. 22:1145-1158. [DOD] [CrossRef]
- Yki-Jarvinen H. Ectopic fat accumulation: an important cause of insulin resistance in humans. J R Soc Med 2002. 95:39-45. [DOD] [CrossRef]
- Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 2002. 23:201-229. [DOD] [CrossRef]
- Bjorntorp P. Adipose tissue distribution and function. Int J Obes 1991. 15:67-81. [DOD]
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003. 144:5159-5165. [DOD] [CrossRef]
- Unger RH, Orci L. Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta 2002. 1585(2-3):202-212. [DOD]
- Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest 1994. 94:2349-2356. [DOD]
- Kelley DE, Williams KV, Price JC, McKolanis TM, Goodpaster BH, Thaete FL. Plasma fatty acids, adiposity, and variance of skeletal muscle insulin resistance in type 2 diabetes mellitus. J Clin Endocrinol Metab 2001. 86:5412-5419. [DOD] [CrossRef]
- Blaak EE. Basic disturbances in skeletal muscle fatty acid metabolism in obesity and type 2 diabetes mellitus. Proc Nutr Soc 2004. 63:323-330. [DOD] [CrossRef]
- Garg A. Acquired and Inherited Lipodystrophies. N Engl J Med 2004. 350:1220-1234. [DOD] [CrossRef]
- Simha V, Garg A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol 2006. 17:162-169. [DOD] [CrossRef]
- Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord 2004. 28:S12-S21. [DOD] [CrossRef]
- Kelley DE. Skeletal muscle fat oxidation: timing and flexibility are everything. J Clin Invest 2005. 115:1699-1702. [DOD] [CrossRef]
- Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. European J Clin Invest 2002. 32:14-23. [DOD] [CrossRef]
- Bollheimer LC, Skelly RH, Chester MW, McGarry JD, Rhodes CJ. Chronic exposure to free fatty acid reduces pancreatic beta cells insulin content by increasing basal insulin secretion that is not compensated for by a corresponding increase in proinsulin biosynthesis translation. J Clin Invest 1998. 101:1094-1101. [DOD]
- Stannard SR, Johnson NA. Insulin resistance and elevated triglyceride in muscle: more important for survival than "thrifty" genes? J Physiol 2004. 554:595-607. [DOD]
- Goodpaster BH, Kelley DE. Skeletal muscle triglyceride: marker or mediator of obesity-induced insulin resistance in type 2 diabetes mellitus? Curr Diab Rep 2002. 2:216-222. [DOD]
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002. 51:2005-2011. [DOD] [CrossRef]
- Sell H, Dietze-Schroeder D, Eckel J. The adipocyte-myocyte axis in insulin resistance. Trends Endocrinol Metab 2006. 17:416-422. [DOD] [CrossRef]
- Menshikova EV, Ritov VB, Toledo FG, Ferrell RE, Goodpaster BH, Kelley DE. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am J Physiol Endocrinol Metab 2005. 288:E818-E825. [DOD] [CrossRef]
- McGarry JD, Dobbins RL. Fatty acids, lipotoxicity and insulin secretion. Diabetologia 1999. 42:128-138. [DOD] [CrossRef]
- Yaney GC, Corkey BE. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 2003. 46:1297-1312. [DOD] [CrossRef]
- Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity. Role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes 2002. 51:S405-S413. [DOD] [CrossRef]
- Dobbins RL, Chester MW, Daniels MB, McGarry JD, Stein DT. Circulating fatty acids are essential for efficient glucose-stimulated insulin secretion after prolonged fasting in humans. Diabetes 1998. 47:1613-1618. [DOD] [CrossRef]
- Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, Polonsky KS. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998. 47:358-364. [DOD] [CrossRef]
- Raman M, Allard J. Non alcoholic fatty liver disease: a clinical approach and review. Can J Gastroenterol 2006. 20:345-349. [DOD]
- Adams LA, Angulo P. Recent concepts in non-alcoholic fatty liver disease. Diabet Med 2005. 22:1129-1133. [DOD] [CrossRef]
- Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab 2006. 91:4753-4761. [DOD] [CrossRef]
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002. 346:1221-1231. [DOD] [CrossRef]
- Roberts EA. Non-alcoholic fatty liver disease (NAFLD) in children. Front Biosci 2005. 10:2306-2318. [DOD] [CrossRef]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 2004. 279:32345-32353. [DOD] [CrossRef]
- Natali A, Ferrannini E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: a systematic review. Diabetologia 2006. 49:434-441. [DOD] [CrossRef]
- Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab 2003. 29(4 Pt 2):6S28-6S35. [DOD]
- Yki-Jarvinen H. Fat in the liver and insulin resistance. Ann Med 2005. 37:347-356. [DOD] [CrossRef]
- Mlinar B, Marc J, Janez A, Pfeifer M. Molecular mechanisms of insulin resistance and associated diseases. Clin Chim Acta 2007. 375:20-35. [DOD] [CrossRef]
- Capeau J, Magre J, Lascols O, Caron M, Bereziat V, Vigouroux C, Bastard JP. Diseases of adipose tissue: genetic and acquired lipodystrophies. Biochem Soc Trans 2005. 33:1073-1077. [DOD] [CrossRef]
- Garg A, Misra A. Lipodystrophies: rare disorders causing metabolic syndrome. Endocrinol Metab Clinic NA 2004. 33:305-331. [DOD] [CrossRef]
- Hegele RA. Phenomics, lipodystrophy, and the metabolic syndrome. Trends Cardiovasc Med 2004. 14:133-137. [DOD] [CrossRef]
- Fasshauer M, Paschke R. Regulation of adipocytokines and insulin resistance. Diabetologia 2003. 46:1594-1603. [DOD] [CrossRef]
- Guzik TJ, Mangalat D, Korbut R. Adipocytokines - novel link between inflammation and vascular function? J Physiol Pharmacol 2006. 57:505-528. [DOD]
- Matsuzawa Y. Adipocytokines and metabolic syndrome. Semin Vasc Med 2005. 5:34-39. [DOD] [CrossRef]
- Faraj M, Lu HL, Cianflone K. Diabetes, lipids, and adipocyte secretagogues. Biochem Cell Bio 2004. 82:170-190. [DOD] [CrossRef]
- Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB. Adiponectin - a key adipokine in the metabolic syndrome. Diabetes Obes Metab 2006. 8:264-280. [DOD] [CrossRef]
- Koerner A, Kratzsch J, Kiess W. Adipocytokines: leptin - the classical, resistin - the controversial, adiponectin - the promising, and more to come. Best Pract Res Clin Endocrinol Metab 2005. 19:525-546. [DOD] [CrossRef]
- Gorden P, Park JY. The clinical efficacy of the adipocyte-derived hormone leptin in metabolic dysfunction. Arch Physiol Biochem 2006. 112:114-118. [DOD] [CrossRef]
- Matsuzawa Y. The metabolic syndrome and adipocytokines. FEBS Lett 2006. 580:2917-2921. [DOD] [CrossRef]
- Lyon CJ, Law RE, Hsueh WA. Minireview: adiposity, inflammation, and atherogenesis. Endocrinology 2003. 144:2195-2200. [DOD] [CrossRef]
- Matsuzawa Y. Therapy Insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nat Clin Pract Cardiovasc Med 2006. 3:35-42. [DOD] [CrossRef]
- Yu YH, Ginsberg HN. Adipocyte signaling and lipid homeostasis: sequelae of insulin-resistant adipose tissue. Circ Res 2005. 96:1042-1052. [DOD] [CrossRef]
- Kaiser N, Nesher R, Donath MY, Fraenkel M, Behar V, Magnan C, Ktorza A, Cerasi E, Leibowitz G. Psammomys obesus, a model for environment-gene interactions in type 2 diabetes. Diabetes 2005. 54:S137-S144. [DOD] [CrossRef]
- Shafrir E, Ziv E, Kalman R. Nutritionally induced diabetes in desert rodents as models of type 2 diabetes: Acomys cahirinus (spiny mice) and Psammomys obesus (desert gerbil). ILAR J 2006. 47:212-224. [DOD]
- Chen D, Wang MW. Development and application of rodent models for type 2 diabetes. Diabetes Obes Metab 2005. 7:307-317. [DOD] [CrossRef]
This article has been cited by other articles:

|
Genetic variation in the G-50T polymorphism of the cytochrome P450 epoxygenase CYP2J2 gene and the risk of younger onset type 2 diabetes among Chinese population: potential interaction with body mass index and family history
Wang CP, Hung WC, Yu TH, Chiu CA, Lu LF, Chung FM, Hung CH, Shin SJ, Chen HJ, Lee YJ
Exp Clin Endocrinol Diabetes 2010. 118(6):346-352
|
|
.gif?preview=preview)
|
Increased epicardial adipose tissue volume in type 2 diabetes mellitus and association with metabolic syndrome and severity of coronary atherosclerosis
Wang CP, Hsu HL, Hung WC, Yu TH, Chen YH, Chiu CA, Lu LF, Chung FM, Shin SJ, Lee YJ
Clin Endocrinol (Oxf) 2009. 70(6):876-882
|
|

|
Measures of coronary artery calcification and association with the metabolic syndrome and diabetes
Ibebuogu UN, Ahmadi N, Hajsadeghi F, Ramirez J, Flores F, Young E, Gopal A, Wong ND, Budoff MJ
J Cardiometab Syndr 2009. 4(1):6-11
|
|
|


)

