Short Review
| Rev Diabet Stud,
2009,
6(2):81-84 |
DOI 10.1900/RDS.2009.6.81 |
The Role of the CXCL10/CXCR3 System in Type 1 Diabetes
Akira Shimada1, Yoichi Oikawa1, Yoshifumi Yamada1, Yoshiaki Okubo1, Shosaku Narumi2
1Department of Internal Medicine, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan
2Department of Molecular Preventive Medicine, University of Tokyo School of Medicine, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113, Japan
Address correspondence to: Akira Shimada, e-mail: asmd@sc.itc.keio.ac.jp
Manuscript submitted July 27, 2009; resubmitted August 3, 2009; accepted August 7, 2009.
Keywords: type 1 diabetes, GAD, T cell, CD4, CD8, T helper cell, Th1, beta-cell proliferation, NOD, insulitis, CD45RB, IL-10, IL-18, IGRP
Abstract
Despite intervention with insulin, type 1 diabetes gradually deteriorates the patients' quality of life. The disease is characterized by an immune-mediated destruction of pancreatic beta-cells. Its etiology, however, remains controversial. Some studies argue that glutamic acid decarboxylase (GAD) antigen and GAD-reactive T cells are critical players in the development of diabetes by affecting the Th cell balance. A T-helper 1 (Th1)-dominant immune response is considered to be important in beta-cell failure in both human and animal models of type 1 diabetes. The Th1-type chemokine, CXCL10, and its receptor, CXCR3, are involved not only in the immune response, but also in the suppression of beta-cell proliferation. Thus, understanding the CXCL10/CXCR3 system may be important for finding a cure. In this short review, we discuss the role of the CXCL10/CXCR3 system in type 1 diabetes and propose relevant treatment options.
Introduction
Type 1 diabetes is an autoimmune disease characterized by the destruction of pancreatic beta-cells. It especially affects the young and the chronic complications reduce their quality of life. To date, there has been no cure for the disease. Even with treatment by islet transplantation, survival of the transplanted islets is less than 5 years in most cases [1]. There is pressing need for a better understanding of the disease pathogenesis and a new treatment strategy based on immune intervention.
The T-helper (Th) cell system is critical for a healthy immune system that balances reactive and suppressive cell compartments. It has been argued that an imbalance in the Th cell system, caused by a predominance of the Th1 response, favors the development of autoimmune diabetes [2]. CXCL10 is an interferon-gamma inducible chemokine and reacts with its receptor, CXCR3, on Th1 cells [3]. Elevated levels of CXCL10 were detected in new onset type 1 diabetes patients and correlated with levels of GAD-reactive CD4 T cells [4]. In addition, CXCL10 is produced by beta-cells, and suppresses beta-cell proliferation [5]. Therefore, we believe that the CXCL10/CXCR3 system plays a decisive role in the pathogenesis of type 1 diabetes. In this short review article, we take a closer look at the CXCL10/CXCR3 system, and discuss its relevance in the disease process and in potential therapy approaches.
The role of the Th1 response in beta-cell failure in the NOD mouse
In the non-obese diabetic (NOD) mouse model, insulitis, with lymphocytic infiltration into the islets, is observed from 5-6 weeks of age. However, this infiltration does not result in rapid onset of diabetes. It takes 3-4 months for the NOD mouse to become diabetic. Therefore, a regulatory mechanism must be at work in this model, at least until the onset of diabetes.
During the early phase of the disease, autoreactive lymphocytes react with insulin, which is why insulin is considered to be a primary autoantigen in type 1 diabetes [6]. Reactivity against insulin results in insulitis, but the occurrence of insulitis does not necessarily mean the onset of diabetes. Diabetes onset may be regulated by a different mechanism. One of the possible mechanisms is a change in the function of regulatory CD4 T cells, (CD45RBlow CD4 cells) during the disease course. In the "regulatory phase", glutamic acid decarboxylase (GAD)-reactive CD45RBlow CD4 cells produce IL-10, which suppresses the onset of diabetes [7]. This GAD-reactive IL-10 response by CD45RBlow CD4 cells balances the polyclonal Th1 response by the same cell type during the disease course. However, it has been reported that the function of CD45RBlow CD4 T cells can change from protective to pathogenic in NOD mice [8-10]. When the Th1 response by CD45RBlow CD4 cells overcomes the GAD-reactive IL-10 response, then diabetes onset can occur [7].
In fact, artificial induction of GAD-reactive T cells in an adoptive transfer system results in the induction of a GAD-reactive IL-10 response and diabetes suppression [11]. This finding supports the view that GAD-reactive responses can be regulatory. On the other hand, the induction of a Th1 response by IL-18 production results in an accelerated diabetes onset [12]. Therefore, the autoimmune Th1 response plays an important role in pancreatic beta-cell failure in type 1 diabetes. However, it is assumed that islet-specific glucose 6 phosphatase catalytic subunit-related protein (IGRP)-reactive CD8 T cells are the "actual killers" of beta-cells [13]. Increased proportions of CD8 T cells in islet lesions have been observed in both animal diabetes models and humans [14, 15]. Therefore, we hypothesize that Th1 cells cooperate with CD8 T cells to destroy beta-cells (Figure 1).
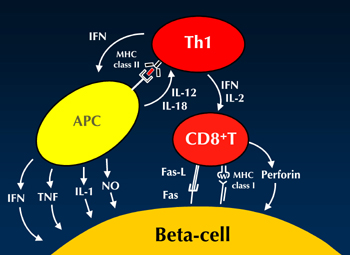 |
 |
Figure 1. Hypothetical illustration of beta-cell destruction. The Th1 cell plays an important role in beta-cell destruction, although CD8 T cells are the actual killers. APC: antigen-presenting cell. IFNγ: interferon gamma. IL: interleukin. TNFα: tumor necrose factor alpha. NO: nitric oxide. |
|
Th1 type chemokine CXCL10 in human type 1 diabetes
A Th1 type chemokine, CXCL10 (also known as IP-10), reacts with its receptor, CXCR3, which is mainly expressed on Th1 cells. These CXCR3-positive cells migrate towards regions of high CXCL10 concentration. Therefore, it has been suggested that CXCL10 could be an important chemokine in type 1 diabetes. A Japanese study [17] reported that serum CXCL10 levels in type 1 diabetes patients, including those experiencing a slow onset disease [16], were significantly higher than that measured in type 2 diabetics and healthy controls. This observation was confirmed in a study with Caucasian patients [18], indicating that this is a consistent phenomenon in type 1 diabetes irrespective of race.
Another interesting observation was that CXCL10 levels correlated with the number of islet antigen-specific CD4 T cells in type 1 diabetics [17, 19, 20]. This suggested that the level of CXCL10 may indicate anti-beta-cell immunity in type 1 diabetes. Also, the serum CXCL10 levels were negatively correlated with disease duration and age of disease onset in type 1 diabetes, suggesting that the CXCL10 level may reflect "disease activity". Thus, serum CXCL10 appears to be a useful marker in type 1 diabetes, and its level may be used to define disease progression.
Role of the CXCL10/CXCR3 system in type 1 diabetes
As mentioned above, serum CXCL10 levels in type 1 diabetics are higher than those in healthy controls, but what does this mean in terms of pathogenesis? In the NOD model of type 1 diabetes, serum CXCL10 levels seem to correlate with high levels of CXCR3 (and high CXCL10) in pancreatic lymph nodes, where T cells are educated [4]. Moreover, longitudinal analysis of serum CXCL10 levels during the disease course indicated that the CXCL10 level seems to predict the onset of diabetes. Taken together, serum CXCL10 levels reflect an accumulation of CXCR3-positive T cells in pancreatic lymph nodes [4], a phenomenon that occurs just before the onset of diabetes.
These data provoke the following question: could the disease course be altered favorably, if the CXCL10/CXCR3 system were blocked by neutralizing CXCL10 antibodies? We tested this hypothesis and found that administration of CXCL10 monoclonal antibodies to a cyclophosphamide-induced NOD diabetes model resulted in a deceleration of diabetes onset [5]. Moreover, induction of anti-CXCL10 antibodies by a gene transfer system in young NOD mice even resulted in disease suppression [21]. These results indicate that blocking the CXCL10/CXCR3 system can be an effective intervention strategy to suppress diabetes onset both in the malignant phase [22], i.e. the active disease phase, and in the benign phase, i.e. the less active phase.
Interestingly, when both CXCL10 and CXCR3 were blocked, no difference in immunological responses, i.e. in cytokine profile, was found. Instead, a difference in pancreatic beta-cell proliferation was discovered. This means that beta-cells were significantly increased by a blockade of the CXCL10/CXCR3 system. It is of note that these interventions were performed before, rather than after, diabetes onset. We do not know, whether or not this approach is similar effective when applied in the hyperglycemic state.
There is evidence that the effect relates to the beta-cell regeneration capacity. It has recently been shown that an interaction between CXCL10 and Toll-like receptor 4 (TLR4) can result in suppression of beta-cell proliferation [23]. As CXCL10 and CXCR3 are co-expressed in pancreatic beta-cells [5], and CXCL10 expression increases as insulitis progresses [4], we believe that, beside its role in the immune response, the CXCL10/CXCR3 system is also critical for the suppression of pancreatic beta-cell proliferation (Figure 2).
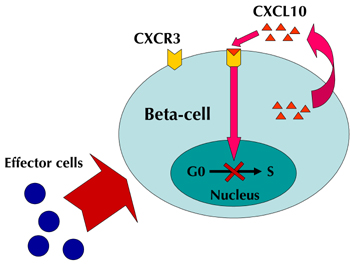 |
|
Figure 2. Suppression of beta-cell proliferation by the CXCL10/CXCR3 system. The destruction of beta-cells by effector cells results in CXCL10 production in beta-cells. CXCL10 binds to CXCR3 on beta-cells, and suppresses beta-cell proliferation in an autocrine manner. Blocking this cycle by CXCL10 neutralizing antibody results in beta-cell proliferation. |
|
Concluding remarks
It is commonly assumed that both immune regulation and beta-cell regeneration are required to cure type 1 diabetes [24]. Some studies have suggested that the CXCL10/CXCR3 chemokine system plays a critical role in the autoimmune process and in beta-cell destruction in type 1 diabetes. Blocking the CXCL10 chemokine in new onset diabetes seems to be a possible approach for treatment. In combination with another regulatory intervention strategy, such as GAD autoantigen sensitization, this approach could contribute to a curative treatment for type 1 diabetes. We envisage that further research into the CXCL10/CXCR3 system will enable to develop a new and effective therapy.
Conflict of interest statement: Dr. Akira Shimada (M.D., Ph.D.) has research grants from Eli Lilly. All other authors declare that they have no conflict of interests.
References
- Shapiro AM, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD, Berney T, Brennan DC, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med 2006. 355(13):1318-1330. [DOD] [CrossRef]
- Sia C. Imbalance in Th cell polarization and its relevance in type 1 diabetes mellitus. Rev Diabet Stud 2005. 2(4):182-186. [DOD] [CrossRef]
- Rotondi M, Lazzeri E, Romagnani P, Serio M. Role for interferon-gamma inducible chemokines in endocrine autoimmunity: an expanding field. J Endocrinol Invest 2003. 26(2):177-180. [DOD]
- Shigihara T, Oikawa Y, Kanazawa Y, Okubo Y, Narumi S, Saruta T, Shimada A. Significance of serum CXCL10/IP-10 level in type 1 diabetes. J Autoimmun 2006. 26(1):66-71. [DOD] [CrossRef]
- Morimoto J, Yoneyama H, Shimada A, Shigihara T, Yamada S, Oikawa Y, Matsushima K, Saruta T, Narumi S. CXC chemokine ligand 10 neutralization suppresses the occurrence of diabetes in nonobese diabetic mice through enhanced beta cell proliferation without affecting insulitis. J Immunol 2004. 173(11):7017-7024. [DOD]
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005. 435(7039):220-223. [DOD] [CrossRef]
- Funae O, Shimada A, Tokui M, Takei I, Saruta T. Balance of GAD65-specific IL-10 production and polyclonal Th1-type response in type 1 diabetes. Autoimmunity 2001. 34(1):45-52. [DOD]
- Shimada A, Rohane P, Fathman CG, Charlton B. Pathogenic and protective roles of CD45RB(low) CD4+ cells correlate with cytokine profiles in the spontaneously autoimmune diabetic mouse. Diabetes 1996. 45(1):71-78. [DOD] [CrossRef]
- Shimada A, Charlton B, Taylor-Edwards C, Fathman CG. Beta-cell destruction may be a late consequence of the autoimmune process in nonobese diabetic mice. Diabetes 1996. 45(8):1063-1067. [DOD] [CrossRef]
- Shimada A, Charlton B, Rohane P, Taylor-Edwards C, Fathman CG. Immune regulation in type 1 diabetes. J Autoimmun 1996. 9(2):263-269. [DOD] [CrossRef]
- Kanazawa Y, Shimada A, Oikawa Y, Okubo Y, Tada A, Imai T, Miyazaki J, Itoh H. Induction of anti-whole GAD65 reactivity in vivo results in disease suppression in type 1 diabetes. J Autoimmun 2009. 32(2):104-109. [DOD] [CrossRef]
- Oikawa Y, Shimada A, Kasuga A, Morimoto J, Osaki T, Tahara H, Miyazaki T, Tashiro F, Yamato E, Miyazaki J, Saruta T. Systemic administration of IL-18 promotes diabetes development in young nonobese diabetic mice. J Immunol 2003. 171(11):5865-5875. [DOD]
- Han B, Serra P, Amrani A, Yamanouchi J, Maree AF, Edelstein-Keshet L, Santamaria P. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med 2005. 11(6):645-652. [DOD] [CrossRef]
- Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009. 155(2):173-181. [DOD] [CrossRef]
- Coppieters KT, von Herrath MG. Histopathology of type 1 diabetes: Old Paradigms and New Insights. Rev Diabet Stud 2009. This issue. [DOD]
- Suzuki R, Shimada A, Maruyama T, Funae O, Morimoto J, Kodama K, Oikawa Y, Kasuga A, Matsubara K, Saruta T, Narumi S. T-cell function in anti-GAD65(+)diabetes with residual beta-cell function. J Autoimmun 2003. 20(1):83-90. [DOD] [CrossRef]
- Shimada A, Morimoto J, Kodama K, Suzuki R, Oikawa Y, Funae O, Kasuga A, Saruta T, Narumi S. Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care 2001. 24(3):510-515. [DOD] [CrossRef]
- Nicoletti F, Conget I, Di Mauro M, Di Marco R, Mazzarino MC, Bendtzen K, Messina A, Gomis R. Serum concentrations of the interferon-gamma-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia 2002. 45(8):1107-1110. [DOD] [CrossRef]
- Shimada A, Kodama K, Morimoto J, Oikawa Y, Irie J, Nakagawa Y, Matsubara K, Maruyama T, Saruta T. Detection of GAD-reactive CD4+ cells in so-called "type 1B" diabetes. Ann N Y Acad Sci 2003. 1005:378-386. [CrossRef]
- Itoh A, Shimada A, Kodama K, Morimoto J, Suzuki R, Oikawa Y, Irie J, Nakagawa Y, Shigihara T, Kanazawa Y, Okubo Y, Motohashi Y, Maruyama T, Saruta T. GAD-reactive T cells were mainly detected in autoimmune-related type 1 diabetic patients with HLA DR9. Ann N Y Acad Sci 2004. 1037:33-40. [DOD] [CrossRef]
- Shigihara T, Shimada A, Oikawa Y, Yoneyama H, Kanazawa Y, Okubo Y, Matsushima K, Yamato E, Miyazaki J, Kasuga A, Saruta T, Narumi S. CXCL10 DNA vaccination prevents spontaneous diabetes through enhanced beta cell proliferation in NOD mice. J Immunol 2005. 175(12):8401-8408. [DOD]
- Yamada S, Irie J, Shimada A, Kodama K, Morimoto J, Suzuki R, Oikawa Y, Saruta T. Assessment of beta cell mass and oxidative peritoneal exudate cells in murine type 1 diabetes using adoptive transfer system. Autoimmunity 2003. 36(2):63-70. [DOD] [CrossRef]
- Schulthess FT, Paroni F, Sauter NS, Shu L, Ribaux P, Haataja L, Strieter RM, Oberholzer J, King CC, Maedler K. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab 2009. 9(2):125-139. [DOD] [CrossRef]
- Okubo Y, Shimada A, Kanazawa Y, Shigihara T, Oikawa Y, Imai T, Miyazaki J, Itoh H. Hyperplastic islets observed in "reversed" NOD mice treated without hematopoietic cells. Diabetes Res Clin Pract 2008. 79(1):18-23. [DOD] [CrossRef]
This article has been cited by other articles:

|
Effect of cholecalciferol as adjunctive therapy with insulin on protective immunologic profile and decline of residual beta-cell function in new-onset type 1 diabetes mellitus
Gabbay MA, Sato MN, Finazzo C, Duarte AJ, Dib SA
Arch Pediatr Adolesc Med 2012. 166(7):601-607
|
|

|
An autoimmune phenotype in vulvar lichen sclerosus and lichen planus: a Th1 response and high levels of microRNA-155
Terlou A, Santegoets LA, van der Meijden WI, Heijmans-Antonissen C, Swagemakers SM, van der Spek PJ, Ewing PC, van Beurden M, Helmerhorst TJ, Blok LJ
J Invest Dermatol 2012. 132(3):658-666
|
|

|
Iloprost for asthma
Luft FC
J Mol Med 2011. 89(1):1-3
|
|

|
CXC chemokine ligand 10 DNA vaccination plus complete Freund's adjuvant reverses hyperglycemia in non-obese diabetic mice
Oikawa Y, Shimada A, Yamada Y, Okubo Y, Katsuki T, Shigihara T, Miyazaki J, Narumi S, Itoh H
Rev Diabet Stud 2010. 7(3):209-224
|
|
|



