Diabetic Perspectives
| Rev Diabet Stud,
2004,
1(4):198-206 |
DOI 10.1900/RDS.2004.1.198 |
Tolerance Induction and Endogenous Regeneration of Pancreatic β-Cells in Established Autoimmune Diabetes
Charles Sia1, Francoise Homo-Delarche2
1Department of Immunology, United Biomedical Inc., 25 Davids Drive, Hauppage, New York 11788, USA.
2CNRS UMR 7059, Paris 7/ D.Diderot University, 2, Place Jussieu, 75005 Paris, France.
Address correspondence to: Charles Sia, e-mail: csia@unitedbiomedical.com
Keywords: type 1 diabetes, tolerance, islet cell regeneration, neogenesis
Abstract
Studies aimed at the understanding of the multifactorial development of autoimmune diabetes have made substantial contributions toward elucidating the molecular mechanisms that open the road to an effective prevention of defective immune responses. Immunomodulatory regimens capable of inducing tolerance are shown to be effective even in the reversal of established autoimmune diabetes in animal models. Experimental trials including the reeducation of autoreactive T cells, depletion of macrophages, dendritic cells, and T cells, as well as the use of monoclonal antibodies, have yielded encouraging results, but have not yet been translated into beneficial clinical outcomes. In addition, we are now seeing an emergence of promising new directions aimed at the induction of islet regeneration by endogenous factors, suggesting that the repair of pancreatic tissue is possible without the need for an engraftment of donor tissue. These recent waves of technological progress have injected new hope for a combined therapy to offer diabetic patients long-term benefits of insulin independence. This article reviews the latest findings on diabetic pathogenesis and discusses promising avenues to tolerance induction and islet regeneration.
Autoimmune diabetes is evoked by a defective immune response
Autoimmune diabetes is hallmarked by the destruction of pancreatic β-cells mediated by cytotoxic T lymphocytes (CTL) and macrophages. However, due to the heterogeneity of the disease and the limited transferability to humans of results obtained in animal models, it is still disputed whether the pathogenic mechanism is conditional on a defective effector or target cell function, or both. Indeed, if an abnormal immune regulation predisposes to autoimmunity, β-cell-specific factors should determine the target of the autoimmune attack. It is generally agreed that a defective function in the immune response results in a loss of tolerance of specific own tissue. Normally, major histocompatibility complex (MHC) class I- and II-restricted immune cells undergo the first stage of maturation in the thymus, where they are educated and normal cellularity is restored by positive selection of T cells that bear properly functioning receptors [1, 2], and continue maturing in the lymph nodes and the periphery. Autoreactive T cells may enter the periphery due to limited or insufficient presentation of self-antigens within the thymus. These T cells further interact with antigen-presenting cells (APCs) in the periphery to become inactivated or deleted in the normal environment [3], which is a critical element of T cell education and results in peripheral tolerance. Education of CTLs is conveyed by the presentation of antigens by MHC class I molecules on cell surfaces of human cells, as well as the selection of T cell progenitors during maturing, which cause mature T cells to recognize self-tissue as such and thus to deter them from becoming autoreactive [4]. Human leukocyte antigen (HLA) genes encode the MHC class I and II surface proteins and thus are important for T cell education and antigen recognition by immunocompetent cells [5]. It is therefore assumed that polymorphisms of these genes lead to different variations of MHC proteins and thus account for over half of the genetic risk of developing type 1 diabetes mellitus (T1DM) [6, 7].
Immune cells are filtered by several checkpoint controls in the thymus and the periphery, suggesting that central as well as peripheral tolerance may be impaired in autoimmune diabetes [8, 9]. Failure of tolerance to self-antigens in the thymus may result from an impaired expression of MHC class I molecules due to a defect in the β2-microglobulin chain which leads to an interrupted presentation of self-antigens to CTLs in NOD mice and humans [10]. The misselected T cells are more susceptible to apoptosis if this failure is combined with a defective activation of NF-κB [11]. Under normal conditions, T cells that encounter self-antigens with high affinity are inactivated and do not leave the thymus [3, 12] or apoptotic programs mediated by TNF-α production are initiated if a cell fails to pass controls [13]. In this context, the loss of tolerance to site-specific tissue is also attributed to a limited antigenic experience of lymphocytes in the thymus and thus such cells are allowed to escape thymic elimination of clonal autoreactive T cells [12, 14-16]. Histological analyses of islet antigens have shown that nonobese diabetic (NOD) mice express markedly reduced levels of the target antigen, ICA69, as compared to non diabetic-prone female B6 and NOD-B6 mice [12]. Peripherally apoptotic programs also exist, but defective Fas-induced antigen-activated apoptosis and clonal expansion of misselected cells may allow autoreactive T cells to survive even peripheral T cell regulation and to enter the pancreas [17-20].
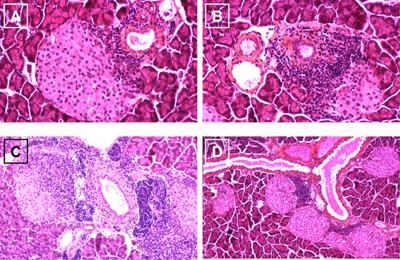
Defective clonal deletion is intrinsic to T cells carrying diabetes susceptibility genes, in particular HLA genes, and thus these cells may be allowed to become specific for islet cells [21]. Such misguided T cells recognize islet antigens, such as GAD and insulin, as foreign and begin to invade the islets of Langerhans and attack insulin-secreting β-cells (Figure 1A). Nevertheless, it should be noted that macrophages and dendritic cells (DCs) are primary contributors to the creation of the autoimmune environment by presenting MHC class II molecules conducive to the development and activation of β-cell-specific Th1-type CD4+ T cells and CD8+ cytotoxic T cells that cause final islet cell destruction in NOD mice and humans [22-24]. In addition, islet antigen-specific autoantibodies are developed by B cells. The process of immune cell infiltration begins early before the clinical manifestation of T1DM. After infiltration, lymphocytes begin to produce pro-inflammatory cytokines that destruct mainly β-cells within the islets of Langerhans (Figures 1B and 1C). In the end, this process results in massive damage to islets, leading to β-cell death and insulin deficiency.
 |
 |
Figure 1. Different stages of insulitis in the pregnant NOD mouse. A: Beginning islet invasion by autoreactive immune cells (purple). B: Augmented T cell presence in the infiltrated islet and beginning β-cell destruction. C: Advanced destruction of β-cells leading to massive damage and lysis. D: Emergence of new islets close to the duct with beginning reinfiltration by autoreactive T cells. |
|
Transient recovery of islet function in the prodromal diabetic stage as a sign of self-regeneration?
Within the first period of insulin therapy after disease onset, a transient recovery of islet function accompanied by a decreased demand of exogenous insulin supply can be observed, known as the 'honeymoon period', before returning to complete insulin-insufficiency. A kind of regeneration or neogenesis of β-cells might occur when the cells are not exposed to glucose-induced stress triggered by hyperglycemia in addition to the inflammatory and/or autoimmune reaction. Such phenomena, together with complete functional recovery, have been observed after experimental induction of endocrine necrosis in the pancreas [25], STZ-induced β-cell death [26], and subtotal pancreatectomy [27], suggesting that pancreatic endocrine tissue is able to renew itself after damage. Physiologically, pregnancy is a well-known model of pancreatic adaptation, resulting in increased β-cell mass that contributes to maternal physiological hyperinsulinemia necessary for placenta and fetal growth. In this regard, Figure 1D shows pregnant nondiabetic 10-week-old NOD mice, with numerous islets in close connection to ducts, as if they have sprouted out of them, a process that is classically observed during islet neogenesis from ductal epithelial precursors. Although most of these islets show lymphocytic infiltration, it can therefore be hypothesized that these new islets either result from residual tissue stemming from the destroyed islets themselves or by neogenesis induced by self-renewal or immature cells. Such cells could be adult stem or progenitor cells that reside within the pancreas or in other organs, such as spleen or blood [28]. Adult stem cells are undifferentiated to some extent, thus not determined to specific tissue [29]. They are able to renew themselves and may become specialized to replace cells that die because of injury or disease [30]. Progenitor cells are also immature but are partly specialized to the tissue in which they reside. It is, however, still questionable if such cells are located within the pancreas of humans with long-term established T1DM. Experimental data suggest that the regenerative potential of the endocrine pancreas diminishes with age [31]. Ongoing lymphocyte attack, however, finally destroys the resprouted islets, such that regeneration is only transient.
Reversal of autoimmunity in established diabetes
Allogeneic islet cell grafts are capable of being effective β-cell surrogates, but are not protected from recurrent autoimmunity, as different immunologic mechanisms are responsible for graft rejection and autoimmunity [32, 33]. The reversal of autoimmunity is thus mandatory for recovering and maintaining of endogenous islet function in established diabetes [34]. As discussed previously, defective immune responses may result from a cascade of events including impaired formation and expression of surface MHC class I proteins and self-antigens, which lead to defects in T cell education and selection [3, 5, 8, 10]. In addition, Hayashi and Faustman found a defective expression of the proteasome β-subunits, the low molecular weight proteins LMP2 and LMP7, in splenocytes of NOD female mice [11]. LMP2 and LMP7 are incorporated in the proteasome that is responsible for intra-cellular protein cleavage and catalysis for antigen presentation, such that T cell selection by antigen presentation functions properly. The proteasome defect induces a defective production and activation of the transcription factor NF-κB that impairs immune and inflammatory responses and increases the susceptibility of misselected T cells to TNF-α mediated apoptosis [35, 11]. This effect can be exploited for abrogating autoreactivity of T cells by the induction of complete Freund’s adjuvant (CFA)-mediated TNF-α production which eliminates these cells and paves the way for the reeducation of newly emerging lymphocytes. This approach has been followed in an experimental protocol by Ryu et al. [32]. CFA administration protected islet grafts in NOD mice that were taken from other NOD mice but not those taken from nondiabetic mice, suggesting that graft vs. host disease and autoimmunity are attributed to different immunologic mechanisms. Simultaneously to CFA administration, naïve host T cells were exposed to donor splenocytes which are unable to express the β2-microglubulin chain and are thus unable to present self-antigens to CTLs. Initially considered as a strategy for bypassing rejection of grafted islets, it has turned out that this intervention may influence the communication among immune cells and lead to a systemic relaxation of islet-specific autoreactivity by the omission of pathogenic clonal events. This regimen resulted in a substantial reversal of autoimmunity and a recreation of β-cell function in about 75-90% of the treated animals in refined experiments [32, 36].
The transferability of results generated in NOD mice to humans is certainly questionable due to the absence of the susceptibility-conferring non-MHC gene locus, IDDM2 on chromosome 11, and the missing identification of the murine homolog of HLA-DR molecules on antigen-presenting cells in mice [37]. Since the human and the murine class II alleles associated with T1DM share conserved features, it can be expected that antigen presentation by APCs is impaired in both humans with T1DM and NOD mice [38], which implies that similar molecular mechanisms are involved in the defect of T cell education in both humans and NOD mice. Therefore, the reversal of impaired antigen presentation in mice presented by Kodama et al. identifies options for reestablishing proper immune communication in diabetic humans, including beneficial T cell education and selection after antigen presentation as well as maintenance of cytokine balance.
Since the proper function of APCs is cardinal for T cell education, autoimmunity may result from the establishment of β-cell specificity by monocytes/macrophages which may be due to immaturity of these cells or defective activation to infiltrate islets [38]. APCs are usually controlled by CD4+CD25+ T cells. Under specific genetic conditions and costimulatory interaction between CD154 and CD40 immature DCs that are able to trigger diabetogenic T cell response are released from suppression by CD4+CD25+ T cells [39]. It is unknown how such T cells attain to the endocrine pancreas. Several assumptions have been tested. Most equivocal is the assumption that the expression of granulocyte macrophage-colony stimulating factor (GM-CSF) in islet cells direct APCs into the islets and activate them to release chemokines and present self-antigens [40]. Recent data show that the macrophage-deficient Csf1op/Csf1op (op/op) mouse phenotypes associated with major insulin mass deficit in fetuses and adults develop abnormal postnatal islet morphogenesis and impaired pancreatic cell proliferation at weaning and late pregnancy [41]. Macrophages are normal constituents of fetal, neonatal and adult pancreas in humans and rodents, capable of secreting various factors involved in islet development and tissue remodeling. It has therefore been hypothesized that their numerous abnormalities, particularly defects in phagocytosis and antigen presentation, observed in autoimmune diabetes, might create a local microenvironment disturbing islet neogenesis and favoring the development of the autoimmune reaction [42]. A beneficial strategy for a reversal of autoimmunity could therefore be to abrogate islet-specificity of immature DCs and macrophages or to suppress their activity. Only few studies have followed this approach; all of them are aimed at the prevention of diabetes in prediabetic subjects by augmenting the activity of protective Th2 cells [43, 44]. This strategy has not yet been tested to reverse established autoimmune diabetes, but could offer potential benefits in restraining APCs from reinducing islet-specific orientation of immune cells.
In order to enhance the number of regulatory CD4+CD25+ T (Treg) cells and therefore to overcome immunosuppressive defects the administration of anti-CD3, monoclonal antibodies (mAb) have been tested [45, 46]. Preclinical data suggest that CD3ε-specific antibodies induce a new population of Treg cells that can lead to a reversal of overt T1DM [47, 48]. Other antibodies directed against costimulatory pathways are only effective at a pre-diabetic stage. Experiments in NOD mice have shown that CTLA4Ig and anti-B7-2 mAb treatment blocked the development of diabetes, but had little effect on the development and severity of insulitis when administered late, at > 10 weeks of age [49].
Another approach aimed at the reversal of established autoimmune diabetes refers to the finding that T cell tolerance to self-antigens may be limited to antigens that are expressed in the thymus and that some pancreatic self-antigens are not (or not sufficiently) expressed in the thymus. Therefore, it has been suggested that the induction of thymic tolerance specific for pancreatic tissue reverses established autoimmune diabetes. Thymic tolerance can be stimulated in animals by exposing immature immune cells with islet cells in the thymus [50].
The effectiveness of insulin administration for tolerizing insulin-primed autoreactive T cells has been tested in a multi-center clinical trial called the Diabetes Prevention Trial-1 (DPT-1). Although the reasons for its failure are not entirely clear at the moment, recent evidence has suggested that intranasal administration does not successfully tolerize autoreactive T cells, but instead induces the generation of diabetogenic cytotoxic T cells. Crippling the immunogenicity of the potential CTL epitopes within the proinsulin molecule has then been shown to induce regulatory T cells capable of mediating the reversal of diabetes in NOD mice [51, 52]. Insulin is at least necessary for concomitant therapy in immunomodulatory or islet regeneration regimens, as it prevents β-cells from glucose-induced and oxidative stress which accompanies diabetes with hyperglycemia via non-enzymatic glycation and alterations in polyol pathway activity [53, 54, 32]. It has been demonstrated in experiments that islet regeneration in formerly STZ-induced hyperglycemic mice is far more effective when insulin is administered concomitantly [26]. Furthermore, the induction of normoglycemia by exogenous insulin supply lowers the susceptibility of islet cells for T cell-mediated infiltration due to the suppression of self-antigens. Macrophages and DCs are the first mediators of islet-specific autoimmunity that present self-antigens to T cells. As β-cell autoantigens are elevated when islets are exposed to hyperglycemia, T-cell infiltration is accelerated by chronic insulin insufficiency [55]. Increased antigen presentation may lead to proliferative responses of islet-antigen-specific autoreactive T cells [56]. Thus T cells appear to strengthen their effort to fight against self-tissue in the presence of an elevated autoantigen level. For this reason it is not advisable to tolerize T cells by antigen-specific vaccination therapy in established autoimmune diabetes. In contrast to the stimulative capacity of islet-derived APCs, splenic APCs appear to show no enhanced potential to stimulate diabetogenic T cells [56]. This is an interesting result in that it confirms the capacity of splenocytes as potential modulators of established autoimmune diabetes.
Regenerative potential of the pancreas
Several approaches to replace damaged pancreatic tissue have been attempted, including the transplantation of allogeneic pancreas [57], allogeneic islet grafts [58], and autologous islet transplantation [59]. Another approach involves the production of insulin-producing tissue and cells by in vitro growth of human β-cells from the epithelium of pancreatic ducts or from acinar cells [60, 61] or by embryonic or adult stem cells [62, 63]. Despite advances in the restoration of the normal insulin function, the surgical process of transplantation is accompanied with severe risks, and is often associated with high morbidity and mortality rates. The problems of graft vs. host disease and a lack of sources arise when donor tissue is used for the replacement of pancreatic insulin-producing cells [64, 65]. Furthermore, immunosuppression applied to avoid graft vs. host disease is associated with adverse effects, such as cancerous affections and islet cell damage [66-68]. The safest strategy would, therefore, be to induce endogenous regeneration.
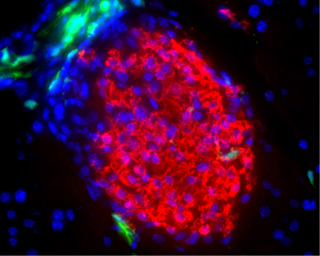
One of the first surprising results of complete pancreatic self-regeneration observed in long-termed insulin-insufficient NOD mice was effectuated by blocking islet-specific autoimmunity [32, 36]. The outcome is relevant in two aspects regarding the recovery of β-cell function in T1DM patients: firstly, allograft-rejection and islet-specific autoimmunity likely result from two distinct mechanisms. The difference in the immune response may be attributed to the antigenic targeting of lymphocytes. While a pancreatic regeneration by native cells leads to the presentation of antigens that induce the reactivation of islet-specific memory T cells, immune ignorance to graft cells is mediated by a different cell-lineage specific for foreign antigens. Secondly, the approach eliminates the need for donor cells and immunosuppressive regimens. The evidence arose from the observation that irradiated splenocytes, used as donor cells, surprisingly led to a similar reconstitution of islets as the use of fresh donor cells. Further examination has shown that allogeneic bone marrow-derived cells from non-diabetes-prone donors are able to initiate endogenous pancreatic tissue regeneration but are largely not participating in the regenerative process [69] (Figure 2).
 |
|
Figure 2. Islet neogenesis without participation of donor cells. Transplanted hematopoietic bone marrow precursors (green) from a diabetes-resistant GFP-transgenic donor mice, genetically marked by green fluorescence protein (GFP), are closely located to the newly formed islet cells (red) but do not directly participate in the regeneration process. Image courtesy of Massimo Trucco, University of Pittsburgh, USA. Reproduction with permission from the Journal of Clinical Investigation. |
|
Induction of islet neogenesis
The capacity of the endogenous pancreas to regenerate itself has been demonstrated in several experiments within the last few years [25, 26, 32, 70]. Current evidence collected from histological studies in animal models suggests that mature islet cells can arise from progenitor cells. During development, such progenitors residing within the bone marrow may differentiate toward a pancreatic β-cell phenotype [71] presumably via an endothelial cell-stage [72, 73] in order to substitute parenchyma cells, which die at the end of their lifespan, at these sites. In adult animals, a phenomenon termed transdifferentiation has been described to suggest islet mature cells may be formed by other cell types; these have included non-insulin secreting cells [74], e.g. acinar cells of the pancreas. A contradictory result concerning the hypothesis of transdifferentiation has been obtained in a recent study by Dor et al. [75]. It is reasoned that the emergence of β-cell regeneration is confined to lineages of β-cell progenitors that have been identified by labeling fully differentiated β-cells expressing the human alkaline phosphatase protein. As the number of labeled β-cells did not change in a defined period of time, the authors conclude that the number of cells derived from non-β-cell progenitors must be approximately zero. The outcome contradicts results of several other studies, where the phenomenon of transdifferentiation has been observed [72, 73] and it is, therefore, doubtful whether the new technical and histological methods that have been applied are unbiased.
Given the evidence that islet cells have regenerative potential, much interest has grown in establishing the mechanisms and conditions of how this could be applied for clinical treatment. Analysis of the regenerating pancreas has led to the discovery that the phenomenon is mediated by a biological active preparation, later named ilotropin [76]. The anti-diabetic property of ilotropin is confirmed by its ability to halt chemically-induced diabetes in hamsters [77]. As mentioned previously, neogenesis may develop from pancreatic ductal cells. Large-scale manufacture of the pancreatic ductal cell-stimulating factor is made with the cloning of a gene encoding for the islet neogenesis associated protein (INGAP). The underlying mechanism is confirmed by the observation that its gene product has the ability to prevent diabetes and convey neogenesis in STZ-treated animals [78, 79].
Pancreatic ductal, endocrine, and exocrine cells are required to undergo differentiation before they are organized to form intact islets. The mechanisms of some of these pathways are becoming increasingly understood. Early stages of pancreatic development are regulated by multiple factors, one of these factors is the homeodomain protein IDX-1 (also known as IPF-1, PDX1 or STF-1). Therefore, it is proposed that α- and β-cells originate from common PDX-1-positive endocrine progenitors [80]. In fact, Seaberg et al. even demonstrate that endocrine pancreatic cells are able to differentiate from a single murine adult pancreatic precursor cell [81]. The origin of this cell line is, however, debatable. It seems neither to be unequivocally an ectodermal, nor a mesodermal or endodermal cell line, as the cells lack specific markers of each of these embryonic cell types. The low frequency and indefinite origin of such precursor and progenitor cells indicate that it will be difficult to identify mechanisms that can be regulated in order to induce β-cell regeneration in vivo.
The differentiation of endocrine cells also involves the multiple roles of several transcription factors belonging to the helix-loop-helix (bHLP) class of homeodomain proteins, while a transcription factor, p48, controls exocrine cell differentiation [82, 83]. A recent study has shown that INGAP in fact mediates the expression of PDX-1 in pancreatic ductal cells and multiple administration of INGAP may reverse diabetes of STZ-treated mice [84]. Apart from INGAP, recent studies have demonstrated that the expression of IDX-1 can also be mediated by another factor, the insulinotropic hormone glucagon-like peptide-1 (GLP-1) produced by the entero-endocrine L-cells of the intestine. Besides the observation in preclinical studies that GLP-1 is capable of restoring normoglycemia in type 2 diabetic mice, the effect of GLP-1 is also attributed to its ability to trigger β-cells secreting insulin together with an accumulation of β-cells within the islets [85].
Conclusion
T1DM is establishing itself as a major autoimmune disease in the human population, especially in Western societies. A more clinically acceptable treatment regimen is therefore needed to effectuate a sustainable and safe cure for the disease in the future. Despite the failure of previous clinical trials attempting to tolerize autoreactive T cells, new advances are being made in this area that hold promise for applications in islet regeneration. Novel discoveries have also been made to understand how islets can be manipulated to undergo neogenesis. These technological developments will undoubtedly add new dimensions to the development of improved treatments for the disease, but there is still a long way to go.
References
- Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell 1987. 49(2):273-280. [DOD] [CrossRef]
- Blackman M, Kappler J, Marrack P. The role of the T cell receptor in positive and negative selection of developing T cells. Science 1990. 248(4961):1335-1341. [DOD]
- Huang FP, MacPherson GG. Continuing education of the immune system - dendritic cells, immune regulation and tolerance. Curr Mol Med 2001. 1(4):457-468. [DOD] [CrossRef]
- Marusic-Galesic S, Stephany DA, Longo DL, Kruisbeek AM. Development of CD4-CD8+ cytotoxic T cells requires interactions with class I MHC determinants. Nature 1988. 333(6169):180-183. [DOD] [CrossRef]
- Mor F, Boccaccio GL, Unger T. Expression of autoimmune disease-related antigens by cells of the immune system. J Neurosci Res 1998. 54(2):254-262. [DOD] [CrossRef]
- Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 1987. 329(6140):599-604. [DOD] [CrossRef]
- Florez JC, Hirschhorn J, Altshuler D. The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu Rev Genomics Hum Genet 2003. 4:257-291. [DOD] [CrossRef]
- Rosmalen JG, van Ewijk W, Leenen PJ. T-cell education in autoimmune diabetes: teachers and students. Trends Immunol 2002. 23(1):40-46. [DOD] [CrossRef]
- Pugliese A, Brown D, Garza D, Murchison D, Zeller M, Redondo M, Diez J, Eisenbarth GS, Patel DD, Ricordi C. Self-antigen-presenting cells expressing diabetes-associated autoantigens exist in both thymus and peripheral lymphoid organs. J Clin Invest 2001. 107(5):555-564. [DOD]
- Faustman D, Li XP, Lin HY, Fu YE, Eisenbarth G, Avruch J, Guo J. Linkage of faulty major histocompatibility complex class I to autoimmune diabetes. Science 1991. 254(5039):1756-1761. [DOD]
- Hayashi T, Faustman DL. Selected contribution: Association of gender-related LMP2 inactivation with autoimmune pathogenesis. J Appl Physiol 2001. 91(6):2804-2815. [DOD]
- Mathews CE, Pietropaolo SL, Pietropaolo M. Reduced thymic expression of islet antigen contributes to loss of self-tolerance. Ann N Y Acad Sci 2003. 1005:412-417. [DOD] [CrossRef]
- Wang J, Lenardo MJ. Molecules involved in cell death and peripheral tolerance. Curr Opin Immunol 1997. 9(6):818-825. [DOD] [CrossRef]
- Mountz JD, Bluethmann H, Zhou T, Wu J. Defective clonal deletion and anergy induction in TCR transgenic lpr/lpr mice. Semin Immunol 1994. 6(1):27-37. [DOD] [CrossRef]
- Oehen SU, Ohashi PS, Burki K, Hengartner H, Zinkernagel RM, Aichele P. Escape of thymocytes and mature T cells from clonal deletion due to limiting tolerogen expression levels. Cell Immunol 1994. 158(2):342-352. [DOD] [CrossRef]
- Hashimoto Y, Maxam AM, Greene MI. T-cell antigen-receptor genes in autoimmune mice. Proc Natl Acad Sci U S A 1986. 83(20):7865-7869. [DOD]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992. 356(6367):314-317. [DOD] [CrossRef]
- Pestano GA, Zhou Y, Trimble LA, Daley J, Weber GF, Cantor H. Inactivation of misselected CD8 T cells by CD8 gene methylation and cell death. Science 1999. 284(5417):1187-1191. [DOD] [CrossRef]
- Arreaza G, Salojin K, Yang W, Zhang J, Gill B, Mi QS, Gao JX, Meagher C, Cameron M, Delovitch TL. Deficient activation and resistance to activation-induced apoptosis of CD8+ T cells is associated with defective peripheral tolerance in nonobese diabetic mice. Clin Immunol 2003. 107(2):103-115. [DOD] [CrossRef]
- Dharnidharka VR, Van Patten Y, Bahjat FR, Clare-Salzler M. Fas stimulation results in selective islet infiltrate apoptosis in situ and reversal of diabetes. Ann N Y Acad Sci 2002. 958:160-162. [DOD]
- Lesage S, Hartley SB, Akkaraju S, Wilson J, Townsend M, Goodnow CC. Failure to censor forbidden clones of CD4 T cells in autoimmune diabetes. J Exp Med 2002. 196(9):1175-1188. [DOD] [CrossRef]
- Dahlen E, Dawe K, Ohlsson L, Hedlund G. Dendritic cells and macrophages are the first and major producers of TNF-alpha in pancreatic islets in the nonobese diabetic mouse. J Immunol 1998. 160(7):3585-3593. [DOD]
- Shinomiya M, Nadano S, Shinomiya H, Onji M. In situ characterization of dendritic cells occurring in the islets of nonobese diabetic mice during the development of insulitis. Pancreas 2000. 20(3):290-296. [DOD] [CrossRef]
- Rosmalen JG, Homo-Delarche F, Durant S, Kap M, Leenen PJ, Drexhage HA. Islet abnormalities associated with an early influx of dendritic cells and macrophages in NOD and NODscid mice. Lab Invest 2000. 80(5):769-777. [DOD]
- Soto C, Mena R, Luna J, Cerbon M, Larrieta E, Vital P, Uria E, Sanchez M, Recoba R, Barron H, Favari L, Lara A. Silymarin induces recovery of pancreatic function after alloxan damage in rats. Life Sci 2004. 75(18):2167-2180. [DOD] [CrossRef]
- Guz Y, Nasir I, Teitelman G. Regeneration of pancreatic beta cells from intra-islet precursor cells in an experimental model of diabetes. Endocrinology 2001. 142(11):4956-4968. [DOD] [CrossRef]
- Hardikar AA, Karandikar MS, Bhonde RR. Effect of partial pancreatectomy on diabetic status in BALB/c mice. J Endocrinol 1999. 162(2):189-195. [DOD] [CrossRef]
- Kodama S, Faustman DL. Routes to regenerating islet cells: stem cells and other biological therapies for type 1 diabetes. Pediatr Diabetes 2004. 5(Suppl 2):38-44. [DOD] [CrossRef]
- Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ. Multi-organ multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 2001. 105:369-377. [DOD] [CrossRef]
- Holtzer H. Cell lineages, stem cells and the 'quantal' cell cycle concept. In: Lord BI, Potten CS, Cole RJ. Stem cells and tissue homeostasis. Cambridge University Press 1978, Cambridge, New York, pp. 1-28. [DOD]
- Tanigawa K, Nakamura S, Kawaguchi M, Xu G, Kin S, Tamura K. Effect of aging on beta-cell function and replication in rat pancreas after 90% pancreatectomy. Pancreas 1997. 15(1):53-59. [DOD]
- Ryu S, Kodama S, Ryu K, Schoenfeld DA, Faustman DL. Reversal of established autoimmune diabetes by restoration of endogenous beta cell function. J Clin Invest 2001. 108(1):63-72. [DOD] [CrossRef]
- Pearson T, Markees TG, Serreze DV, Pierce MA, Marron MP, Wicker LS, Peterson LB, Shultz LD, Mordes JP, Rossini AA, Greiner DL. Genetic disassociation of autoimmunity and resistance to costimulation blockade-induced transplantation tolerance in nonobese diabetic mice. J Immunol 2003. 171(1):185-195. [DOD]
- von Herrath M, Homann D. Islet regeneration needed for overcoming autoimmune destruction - considerations on the pathogenesis of type 1 diabetes. Pediatr Diabetes 2004. 5(Suppl 2):23-28. [DOD] [CrossRef]
- Hayashi T, Faustman D. NOD mice are defective in proteasome production and activation of NF-kappaB. Mol Cell Biol 1999. 19(12):8646-8659. [DOD]
- Kodama S, Kuhtreiber W, Fujimura S, Dale EA, Faustman DL. Islet regeneration during the reversal of autoimmune diabetes in NOD mice. Science 2003. 302(5648):1223-1227. [DOD] [CrossRef]
- Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 1999. 5(6):601-604. [DOD]
- Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet 1995. 345(8948):491-492. [DOD] [CrossRef]
- Serra P, Amrani A, Yamanouchi J, Han B, Thiessen S, Utsugi T, Verdaguer J, Santamaria P. CD40 ligation releases immature dendritic cells from the control of regulatory CD4+CD25+ T cells. Immunity 2003. 19(6):877-889. [DOD] [CrossRef]
- Krakowski M, Abdelmalik R, Mocnik L, Krahl T, Sarvetnick N. Granulocyte macrophage-colony stimulating factor (GM-CSF) recruits immune cells to the pancreas and delays STZ-induced diabetes. J Pathol 2002. 196(1):103-112. [DOD] [CrossRef]
- Banaei-Bouchareb L, Gouon-Evans V, Samara-Boustani D, Castellotti MC, Czernichow P, Pollard JW, Polak M. Insulin cell mass is altered in Csf1op/Csf1op macrophage-deficient mice. J Leukoc Biol 2004. 76(2):359-367. [DOD] [CrossRef]
- Homo-Delarche F, Drexhage HA. Immune cells, pancreas development, regeneration and type 1 diabetes. Trends Immunol 2004. 25(5):222-229. [DOD] [CrossRef]
- Feili-Hariri M, Falkner DH, Morel PA. Regulatory Th2 response induced following adoptive transfer of dendritic cells in prediabetic NOD mice. Eur J Immunol 2002. 32(7):2021-2030. [DOD] [CrossRef]
- Ma L, Qian S, Liang X, Wang L, Woodward JE, Giannoukakis N, Robbins PD, Bertera S, Trucco M, Fung JJ, Lu L. Prevention of diabetes in NOD mice by administration of dendritic cells deficient in nuclear transcription factor-kappaB activity. Diabetes 2003. 52(8):1976-1985. [DOD]
- Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, Ten S, Sanz M, Exley M, Wilson B, Porcelli S, Maclaren N. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest 2002. 109(1):131-140. [DOD] [CrossRef]
- Herold KC, Taylor L. Treatment of Type 1 diabetes with anti-CD3 monoclonal antibody: induction of immune regulation? Immunol Res 2003. 28(2):141-150. [DOD]
- Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 2003. 9(9):1202-1208. [DOD] [CrossRef]
- Chatenoud L. CD3 antibody treatment stimulates the functional capability of regulatory T cells. Novartis Found Symp 2003. 252:279-286. [DOD]
- Lenschow DJ, Ho SC, Sattar H, Rhee L, Gray G, Nabavi N, Herold KC, Bluestone JA. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med 1995. 181(3):1145-1155. [DOD] [CrossRef]
- Herold KC, Montag AG, Buckingham F. Induction of tolerance to autoimmune diabetes with islet antigens. J Exp Med 1992. 176(4):1107-1114. [DOD] [CrossRef]
- Hanninen A, Harrison LC. Mucosal Tolerance to Prevent Type 1 Diabetes: Can the Outcome Be Improved in Humans? Rev Diabetic Stud 2004. 1(3):113-121. [DOD]
- Martinez NR, Augstein P, Moustakas AK, Papadopoulos GK, Gregori S, Adorini L, Jackson DC, Harrison LC. Disabling an integral CTL epitope allows suppression of autoimmune diabetes by intranasal proinsulin peptide. J Clin Invest 2003. 111:1365-1371. [DOD] [CrossRef]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003. 52(1):1-8. [DOD]
- Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka TA. Involvement of Oxidative Stress and the JNK Pathway in Glucose Toxicity. Rev Diabetic Stud 2004. 1(4):163-172. [DOD]
- Yoon JW, Jun HS. Cellular and molecular pathogenic mechanisms of insulin-dependent diabetes mellitus. Ann N Y Acad Sci 2001. 928:200-211. [DOD]
- Judkowski V, Krakowski M, Rodriguez E, Mocnick L, Santamaria P, Sarvetnick N. Increased islet antigen presentation leads to type-1 diabetes in mice with autoimmune susceptibility. Eur J Immunol 2004. 34(4):1031-1040. [DOD] [CrossRef]
- Robertson RP, Sutherland DE, Kendall DM, Teuscher AU, Gruessner RW, Gruessner A. Metabolic characterization of long-term successful pancreas transplants in type 1 diabetes. J Invest Med 1996. 44:549-555. [DOD]
- Hering BJ, Browatzki CC, Schultz A, Bretzel RG, Federlin KF. Clinical islet transplantation - registry report, accomplishments in the past and future research needs. Cell Transplant 1993. 2:269-282. [DOD]
- Pyzdrowski KL, Kendall DM, Halter JB, Nakhleh RE, Sutherland DE, Robertson RP. Preserved insulin secretion and insulin independence in recipients of islet autografts. N Engl J Med 1992. 327:220-226. [DOD]
- Bonner-Weir S, Taneja M, Weir G, Tatarkiewicz K, Song K, Sharma A, O'Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A 2001. 97:799-800. [DOD]
- Street CN, Lakey JRT, Rajotte RV, Shapiro AMJ, Kieffer TJ, Lyon JG, Kin T, Korbutt GS. Enriched Human Pancreatic Ductal Cultures Obtained from Selective Death of Acinar Cells Express Pancreatic and Duodenal Homeobox Gene-1 Age-Dependently. Rev Diabetic Stud 2004. 1:66-79. [DOD] [CrossRef]
- Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL. Insulin production by human embyronic stem cells. Diabetes 2001. 50:1-7. [DOD]
- Lechner A, Nolan AL, Blacken RA, Habener JF. Redifferentiation of insulin-secreting cells after in vitro expansion of adult human pancreatic islet tissue. Biochem Biophys Res Commun 2005. 327(2):581-588. [DOD] [CrossRef]
- Schmidt P, Krook H, Maeda A, Korsgren O, Benda B. A new murine model of islet xenograft rejection: graft destruction is dependent on a major histocompatibility-specific interaction between T-cells and macrophages. Diabetes 2003. 52(5):1111-1118. [DOD]
- Li H, Ricordi C, Inverardi L. Effects of graft-versus-host reaction on intrahepatic islet transplants. Diabetes 1999. 48(12):2292-2299. [DOD]
- Papaconstantinou HT, Sklow B, Hanaway MJ, Gross TG, Beebe TM, Trofe J, Alloway RR, Woodle ES, Buell JF. Characteristics and survival patterns of solid organ transplant patients developing de novo colon and rectal cancer. Dis Colon Rectum 2004. 47(11):1898-1903. [DOD] [CrossRef]
- Trofe J, Beebe TM, Buell JF, Hanaway MJ, First MR, Alloway RR, Gross TG, Woodle ES. Posttransplant malignancy. Prog Transplant 2004. 14(3):193-200. [DOD]
- Drachenberg CB, Klassen DK, Weir MR, Wiland A, Fink JC, Bartlett ST, Cangro CB, Blahut S, Papadimitriou JC. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation 1999. 68(3):396-402. [DOD] [CrossRef]
- Zorina TD, Subbotin VM, Bertera S, Alexander AM, Haluszczak C, Gambrell B, Bottino R, Styche AJ, Trucco M. Recovery of the endogenous beta cell function in the NOD model of autoimmune diabetes. Stem Cells 2003. 21(4):377-388. [DOD] [CrossRef]
- George M, Ayuso E, Casellas A, Costa C, Devedjian JC, Bosch F. Beta cell expression of IGF-I leads to recovery from type 1 diabetes. J Clin Invest 2002. 109(9):1153-1163. [DOD] [CrossRef]
- Ianus A, Holz GG, Theise ND, Hussain MA. In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J Clin Invest 2003. 111:843-850. [DOD] [CrossRef]
- Zulewski H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Muller B, Vallejo M, Thomas MK, Habener JF. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001. 50:521-533. [DOD]
- Bonner-Weir S, Baxter L A, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 1993. 42:1715-1720. [DOD]
- Song KH, Ko SH, Ahn YB, Yoo SJ, Chin HM, Kaneto H, Yoon KH, Cha BY, Lee KW, Son HY. In vitro transdifferentiation of adult pancreatic acinar cells into insulin-expressing cells. Biochem Biophys Res Commun 2004. 316(4):1094-1100. [DOD] [CrossRef]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004. 429(6987):41-46. [DOD] [CrossRef]
- Pittenger GL, Rosenberg L, Vinik AI. Partial purification and characterization of ilotropin, a pancreatic-islet specific growth factor. J Cell Biol 1991. 115:270A. [DOD]
- Rosenberg L, Vinik AI, Pittenger GL. Islet regeneration in the diabetic hamster pancreas with restoration of normoglycemia can be induced by a local growth factor(s). Diabetologia 1996. 39:256-262. [DOD]
- Rafaeloff R, Pittenger GL, Barlow SW, Qin XF, Yan B, Rosenberg L, Duguid WP, Vinik AI. Cloning and sequencing of the pancreatic islet neogenesis asosciated protein (INGAP) genes and its expression in islet neogenesis in hamsters. J Clin Invest 1997. 99:2100-2109. [DOD]
- Tam J, Rosenberg L, Maysinger D. INGAP peptide improves nerve function and enhances regeneration in streptozotocin-induced diabetic C57BL/6 mice. FASEB J 2004. 18(14):1767-1769. [DOD]
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000. 127(11):2317-2322. [DOD]
- Seaberg RM, Smukler SR, Kieffer TJ, Enikolopov G, Asghar Z, Wheeler MB, Korbutt G, van der Kooy D. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat Biotechnol 2004. 22(9):1115-1124. [DOD] [CrossRef]
- Sander M, German MS. The cell transcription factors and the development of the pancreas. J Mol Med 1997. 75:327-340. [DOD]
- Krapp A, Knofler M, Ledermann B, Burki K, Berney C, Zoerkler N, Hagenbuchle O, Wellauer PK. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev 1998. 12(23):3752-3763. [DOD]
- Rosenberg L, Lipsett M, Yoon JW, Prentki M, Wang R, Jun HS, Pittenger GL, Taylor-Fishwick D, Vinik AI. A pentadecapeptide fragment of islet neogenesis-associated protein increases beta-cell mass and reverses diabetes in C57BL/6J mice. Ann Surg 2004. 240(5):875-884. [DOD] [CrossRef]
- Stoffers DA, Kieffer TJ, Hussain MA, Drucker DJ, Bonner-Weir S, Habener JF, Egan JM. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes 2000. 49(5):741-748. [DOD]
|


)
)

