Editorial
| Rev Diabet Stud,
2004,
1(4):156-164 |
DOI 10.1900/RDS.2004.1.156 |
Differential Roles of Costimulatory Signaling Pathways in Type 1 Diabetes Mellitus
Bernhard O. Boehm1, Jeffrey A. Bluestone2
1Division of Endocrinology and Diabetes, Department of Internal Medicine, University of Ulm, Germany
2Diabetes Center, Department of Medicine, University of California San Francisco, San Francisco, CA 94143, USA.
Address correspondence to: Bernhard O. Boehm, e-mail: bernhard.boehm@medizin.uni-ulm.de
Introduction
Insulin-dependent type 1 diabetes mellitus (T1DM) is a chronic immune mediated disease in which T lymphocytes recognizing pancreatic islet cell antigens play a major role in the disease process [2, 3]. It has become increasingly clear that autoreactive T cells are important in the development and progression of this disease. The initial event in the pathogenesis of autoimmune disease is thought to be the priming of naïve autoreactive T cells, perhaps as a consequence of environmental exposure to shared epitopes (antigenic mimicry) or generalized inflammation/innate immunity [2-4].
Maximum activation of naïve T lymphocytes requires at least two independent signals. The first, an antigen-specific signal is sent via the clonotypic T cell receptor (TCR) which recognizes the appropriate peptide-MHC complex. The second signal, termed costimulatory signal, is independent of the antigen receptor and is critical to allow full activation. In addition, costimulatory signaling controls proliferation, prevents anergy and/or apoptosis, and induces differentiation to effector function and memory status. Costimulatory signals also regulate inhibitory receptors upon lymphocyte activation in a time-dependent manner [5, 6]. It is now clear that the delicate balance of these positive and negative regulatory pathways underpins the outcome - self-tolerance versus autoimmunity [7].
Therefore, the immune response observed in T1DM seems to be related to an imbalance of the sensitively regulated process which usually controls the activation of thymus-dependent lymphocytes. In this article, we summarize the role of costimulation in the development and progression of T1DM. In addition to the basic biology, we highlight important clinical advances in costimulation blockade that may impact this disease.
Costimulation - help is critical for T cell activation
It has long been recognized that T lymphocytes stimulated through the antigen receptor alone fail to produce cytokines, are unable to sustain proliferation, or may become unresponsive to a subsequent stimulation [5]. Lack of costimulation often leads to T cell apoptosis due to underexpression of pro-survival factors such as IL-2 and bcl-xL [8]. Recently it was found that interactions between receptor/ligand pairs of cell surface molecules on the responder lymphocyte and an antigen-presenting cell (APC) are critical for T cell activation [5, 9]. In addition, there is growing evidence for a more sophisticated interaction, i.e. a highly complex bidirectional communication between APC and T cells. This crosstalk allows a specific fine-tuning of lymphocyte activation.
The specialized junction between a T lymphocyte and an antigen-presenting cell, the immunological synapse, consists of a central cluster of T cell receptors surrounded by a ring of adhesion and costimulatory molecules. Immunological synapse formation has been shown to be an active and dynamic mechanism that allows T cells to distinguish potential antigenic ligands. This complex cellular structure that forms at the interface of a T cell and a cell that expresses the appropriate peptide-MHC complexes resembles neuronal interfaces with an ongoing cell-to-cell communication, finally leading to T cell activation or a putative modulation of T cell activities [9, 10].
Candidate costimulatory molecules
To act as a costimulatory molecule, the receptor has to fulfill the following functional criteria: (a) the molecule must initiate a positive signal without simply increasing TCR avidity or (b) by enhancing the recruitment of tyrosine kinases to the TCR complex, as in the case of the accessory molecules CD4 and CD8, which act as coreceptors. The most prominent cell surface molecule shown to function as a costimulatory receptor is CD28 [8, 11]. CD28 is a disulfide-linked homodimer and is constitutively expressed on most T-lineage cells. Since the identification of CD28, the number of proposed costimulatory molecules has grown significantly. CD28 binding to its ligands, CD80 (B7.1) and CD86 (B7.2), has been shown to enhance a variety of secondary biochemical signaling pathways resulting in enhanced cell survival, proliferation and differentiation (Figure 1). However, there are numerous examples of immune responses occurring in CD28-deficient settings suggesting that additional T cell surface receptors [12], such as TNF family members CD154 (CD40L) and OX40, can serve similar costimulatory functions [13, 14]. Moreover, in addition to the role of costimulation in the initiation of a primary immune response, a number of activation-dependent receptor-ligand pairs have been implicated as "amplifying" costimulatory pathways involved in ongoing immune responses [15].
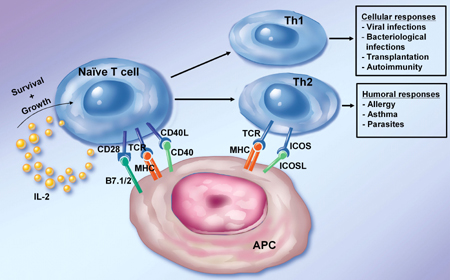 |
 |
Figure 1. Role of costimulation in T cell activation. The figure depicts the role of various costimulatory molecules in the activation of naïve and differentiated T cells. Naïve T cells and Th1 cells rely heavily on CD28 and CD40L engagement to expand and differentiate. The ICOS/ICOSL pathway works on activated T cells with an especially potent signal to promote Th2 development. The activated T helper cell subsets finally promote different ranges of cellular and humoral immune responses. |
|
Costimulatory molecules can be divided into two broad classes based on sequence homologies. The first class contains the related CD28 and inducible costimulator (ICOS) molecules. CD28 and ICOS are both disulfide-linked homodimers that bind to distinct members of the B7 family of surface proteins. CD28 uses a highly conserved motif in the CDR3-like loop for binding to the B7 molecules. The ICOS molecule has recently attracted attention as an alternative costimulatory molecule. ICOS is structurally related to CD28 but does not detectably bind to B7.1 or B7.2. Instead, the ligand for ICOS (ICOSL) is a novel B7 family member. ICOS is either absent or expressed at very low levels on naïve T cells, but it is up-regulated upon stimulation. ICOS/ICOSL interactions therefore optimize and further differentiate responses of recently activated Th cells (Figure 1) [16]. ICOS costimulation has been shown to contribute to the production of various effector cytokines (IFN-γ, TNF-α, IL-4, IL-5, and IL-10). Thus, CD28 and ICOS appear to be major costimulatory molecules for the activation of T cells, with naïve and resting T cells using CD28 and activated and effector T cells using ICOS.
Antigen-presenting cells show differential regulation of B7.1, B7.2, and ICOSL, allowing specificity in eliciting responses from naïve versus activated (or memory) T cells. Most importantly, however, ICOSL expression can be induced in fibroblasts in culture, and nonlymphoid tissues in mice, by treatment with the inflammatory agents LPS and TNF-α, whereas CD28 ligands are restricted to cells of the leukocyte lineages and are not induced in stromal tissues by similar treatment [17]. Thus, inflamed peripheral tissues may selectively stimulate antigen-experienced T cells, while naïve T cells are primed locally in lymphoid tissues draining the site of antigen expression. This observation may provide a clue for an in situ regulation of already preformed (autoreactive) T cell responses. CD28 and ICOS, acting at different stages of T cell immune responses, are required for proper Th cell activation, differentiation, and effector cytokine expression.
Two other receptors, cytotoxic lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1), share structural homology with this class and also bind B7 family members. CTLA-4 is 50% homologus to CD28 in its extracellular domain and binds the same B7.1 and B7.2 ligands using a conserved amino acid sequence (MYPPPY) in its CDR3 domain [18]. CTLA-4 inhibits effector T cell activation by a combination of competition for CD28 ligation as well as a biochemical-mediated dephosphorylation of the T cell receptor complex based on co-localization of tyrosine and serine/threonine phosphatase activity within the immunologic synapse [19-22] (Figure 2). PD-1, a CD28 homologue, contains two immunoreceptor tyrosine-based motifs that are phosphorylated upon receptor engagement and recruit Src homology 2-domain-containing tyrosine phosphatase 2. Two novel B7 family members, programmed death-1 ligand (PD-L)1 (B7-H1) and PD-L2 (B7-DC), have been shown to down-regulate T cell activation through their mutual receptor, programmed death-1 (PD-1) (Figure 2). The impact of the negative regulatory effects of these two pathways should not be underestimated, as mice deficient in PD-1 develop autoimmune disorders suggesting a defect in peripheral tolerance [23-25] and CTLA-4-deficient mice die within 2-4 four weeks of age from an uncontrolled lymphoproliferation [26-28]. Thus, it has only recently been fully appreciated that there is an intimate and regulated response to antigenic stimuli such that the relative expression of these various receptors and their targets can either tip the balance towards immunity or tolerance.
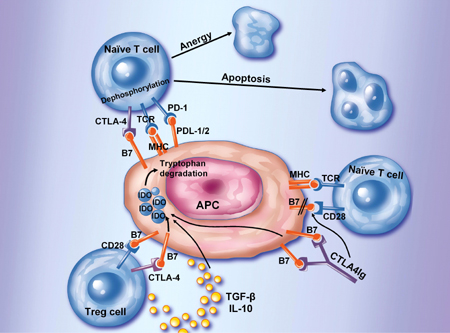 |
|
Figure 2. Inhibition of T cell activation. A number of cell surface molecules controls and suppresses T cell activation by directly suppressing T cell activation (via CTLA-4 or PD-1), by directly signaling the T cell, or indirectly in the case of CTLA4Ig, or regulatory T cells by binding to DCs inducing IDO. IDO catalyzes the degradation of tryptophan to kynureine and subsequent catabolic byproducts. Local decreases in the availability of tryptophan and the presence of its catabolic byproducts inhibit T cell proliferation and may induce apoptotic death. Inhibition of clonal expansion leads to a functional state of tolerance. This pathway may be accentuated by the presence of IL-10 or TGF-β. Regulatory T cells secretion of IL-10 may induce these B7-dependent DC-signaling pathways as well. |
|
The second class of costimulatory receptors are members of the TNF receptor (TNFR) family. These include CD40, the major B cell costimulatory molecule, as well as OX-40, 4-1BB, CD30, and CD27. The ligands for these receptors are membrane-bound members of the TNF family [29]. These members of the TNF receptor family will not be further addressed in this article.
IDDM genes
Complex autoimmune diseases like T1DM reveal a multigenic genetic susceptibility with more than one gene involved. The T1DM susceptibility genes are not mutated, they do not represent rare altered variants from deleterious mutations; instead they are polymorphic [30, 31]. Using a genome-wide scanning approach, a large number of diabetes-associated genes have been described. The IDDM12 locus located on chromosome 2q33 contains the CTLA-4, ICOS and CD28 genes. Further studies have unambiguously linked costimulatory receptors to the T1DM susceptibility gene locus [32].
The CTLA-4 gene is the strongest candidate gene for autoimmune diseases in this region, based on both the biological and genetic studies. Polymorphisms have been described and found to have preferential transmission to the affected siblings in segregation analysis. Most importantly, in a multi-ethnic analysis (U.S. Caucasian, Mexican-American, French, Spanish, Korean, and Chinese), collection of both simplex and multiplex families was typed for 10 polymorphic markers within a genomic interval of approximately 300 kb, which contained the candidate genes CTLA4, ICOS and CD28. The consideration of ethnic and racial representations in these studies provided additional information for a more general role of the gene polymorphisms related to T1DM [33, 34].
Transmission disequilibrium tests (TDT) revealed significant association/linkage with three markers within CTLA-4 and two immediate flanking markers (D2S72 and D2S105) on each side of CTLA-4 but not with more distant markers including the candidate gene CD28. These observations in this setting, as well as similar linkages in Graves’ disease [35], strongly support the conclusion that the CTLA-4 gene is a major susceptibility gene for T1DM in various populations and of functional importance in different environmental settings.
IDDM susceptibility genes become functional
Further studies mapped disease susceptibility to a polymorphism in the non-coding 6.1 kb 3’ end associated with lower messenger RNA levels of a soluble form of CTLA-4 resulting from alternative splicing [32, 36]. This soluble form, which results from a variant CTLA-4 molecule, may control immune functions in a variety of ways including the regulation of CD28 costimulation; regulatory T cell function; induction of indoleamine-2,3-dioxygenase (IDO) and direct effects on membrane-bound CTLA-4 function (Figure 2). A polymorphism at position 49 of exon 1 (known as G/G variant) was found to be associated with decreased expression of the variant CTLA-4 molecule [35, 37, 38]. A relationship between genotype and the expression of the CTLA-4 isoforms has been found. Like in humans, type 1 diabetes susceptibility in the nonobese diabetes (NOD) mouse model is associated with a CTLA-4 gene polymorphism. In this spontaneous disease setting, the splice variant results in a reduced production of a cell surface molecule lacking the CD80/CD86 ligand-binding domain [32, 36]. Genome scanning in humans and the NOD mouse model has therefore contributed significantly to identify pathways in a complex (autoimmune) disorder.
Functional studies in T cells have provided evidence that the alternatively spliced form of CTLA-4, named ligand-independent CTLA-4 (liCTLA-4), lacks exon 2 including the MYPPPY motif which is essential for binding to the costimulatory ligands B7.1 and B7.2. In in vitro studies it also became clear that liCTLA-4 expressed as a protein in primary T cells and strongly inhibited T cell responses by binding and dephosphorylating the TcRζ chain. The expression levels of liCTLA-4 were higher in memory/regulatory T cells from diabetes-resistant NOD congenic mice compared to susceptible NOD mice indicating that the spliced form liCTLA-4 is critical for the down-regulation of T cell-mediated autoimmunity [36].
Experimental evidence show an inhibitory role for this novel isoform of CTLA-4, as expression of the cytoplasmic domain of liCTLA-4 alone is sufficient to deliver a negative signal even though it lacks an extra-cellular domain required for interaction with B7 molecules. Thus, CTLA-4 isoforms may function to increase the activation threshold of autoreactive T cells and may therefore have an important role in various autoimmune disorders.
Recently structural requirements for T cell costimulation revealed unusual modes of dimerization for the CTLA-4 costimulatory receptor and its B7 ligands. These distinctive quaternary structures potentially endow both receptor and ligand with bivalent binding properties. Most probably these members of the costimulatory receptor-ligand family may potentially form a highly ordered, alternating network of CTLA-4 and B7 homodimers that may represent the organization of these molecules and their associated signaling partners within the immunological synapse. One might postulate that CTLA-4 isoforms may alter these structural features and thus lead to a modification of signaling [39].
Disease models using costimulatory signals
The critical role of costimulatory signaling has been demonstrated initially in genetically disrupted CTLA-4 knockout mice that develop polyclonal lymphocyte activation and proliferation, leading to massively enlarged lymph nodes as well as spleen and fatal multiorgan lymphocytic infiltrations. Transgenic approaches demonstrated that RIP-mCD80+ mice, which express the mouse costimulatory molecule B7.1 (mCD80) on pancreatic β-cells under the control of the rat insulin-1 promoter, were extraordinarily susceptible to autoantigen-induced diabetes, while spontaneous disease was rare. However, multiple low dose injections of the β-cell toxin streptozotocin in RIP-mCD80+ β-cells resulted in T-cell-mediated insulin-dependent diabetes mellitus in RIP-mCD80+ mice suggesting that diabetes induction involved both Ag-specific recognition of self-target molecules in the inflamed pancreatic islet (signal 1) and CD28 costimulation (signal 2) [40].
The breaking of "horror autotoxicus" in RIP-mCD80+ mice which co-express to the pancreatic β-cell-expressed glycoprotein (GP) of the lymphocytic choriomeningitis virus (LCMV), was elicited by a single injection of syngeneic fibroblastoid cell lines (FCL) loaded with the immunodominant LCMV-GP peptide, gp33. While both RIP-GP+ and RIP-CD80+GP+ mice mounted moderate CD4-independent CTL responses, only CD80+GP+ mice developed severe insulitis. Diabetes was evoked due to islet-infiltration of activated, gp33-specific, CD8+ T-cells. These T cells secreted large amounts of IFN-γ. DNA immunization using plasmids encoding LCMV-GP or murine preproinsulin also efficiently induced Ag-specific RIP-CD80-dependent diabetes [41]. Therefore, aberrant CD80-expression in a peripheral tissue, which can be induced by various "non-specific" signals to the innate immune system, may disrupt the tissue's natural resistance to CD8+ T-cell-mediated autoimmune destruction [42]. Similar effects were observed following local co-expression of TNF with human B7.1 in the islets, resulting in a profound and rapid induction of diabetes mediated largely by CD8+ cytolytic T cells.
Genetic vaccination with (pro)insulin was able to adversely trigger autoimmune diabetes in two mouse models of type 1 diabetes, using intramuscular DNA vaccination for antigen administration. In female non-obese diabetic (NOD) mice, diabetes development was enhanced after preproinsulin (ppIns) DNA treatment, and natural diabetes resistance in male NOD mice was diminished by ppIns DNA vaccination. GAD65 DNA conferred partial diabetes protection, whereas empty DNA plasmid was without effect. In RIP-B7.1 C57BL/6 mice, which provides a setting of expressing the T cell costimulatory molecule B7.1 in pancreatic β-cells against a non-diabetogenic background, again autoimmune diabetes occurred in 70% of animals after ppIns vaccination. Diabetes did not develop spontaneously in RIP-B7.1 mice or after GAD65 or control DNA treatment. Diabetes was characterized by a CD4+CD8+ T cell infiltration of pancreatic islets and severe insulin deficiency [43].
Overall, these functional studies showed that up-regulation of costimulatory pathways is an important component of normal immune homeostasis. Expression of B7.1 in the target tissue dramatically alters the cellular and molecular requirements for the development of autoimmunity [42, 43]. Experimental data in animal models also showed that potentially autoreactive T cells that have escaped negative selection in the thymus must be strictly controlled in the periphery to avoid autoimmune disease and that they can escape such a control by the help of impairment of costimulatory signaling.
Perhaps the most unexpected observation was the finding that disruption of the CD28/B7 pathway in the NOD mouse strain using either mAb or knock technologies resulted in exacerbation of the disease [12]. This paradoxical observation supported a more complex role of this pathway in the autoimmune disease setting. In fact, there is now substantial evidence that CD28 is as important in supporting the development and survival of CD4+CD25+ regulatory T cells (Tregs) that are essential suppressors of disease in the NOD mouse [7] (Figure 2). The disruption of the CD28 pathway results in the complete loss of functional regulatory T cells and the residual pathogenic cells are sufficient to mediate disease. As importantly, individual B7.1 versus B7.2 knockout mice have different effects on disease outcome. In both cases the disruption of each molecule individually has minimal effect on Tregs. In the case of B7.1-deficiency disease incidence is exacerbated while B7.2 deficiency totally abrogates diabetes [44]. The results can be interpreted as follows: Under conditions of normal Treg numbers, B7.2 is the prominent CD28 ligand involved in T cell pathogenesis while B7.1 is the preferred ligand for CTLA-4 leading to the yin-yang in this system [45-48].
Finally, there is increasing evidence that the function of the CTLA-4 molecule is also more complex. As stated above, there is no doubt that CTLA-4 can directly regulate T cell activation by attenuating TCR signaling. However, several groups have also implicated CTLA-4 in Treg function [49-51]. The mechanism is unclear, but may be mediated through TGFβ-production by Tregs and induction of the suppressive tryptophan metabolic enzyme (IDO) by Tregs in APCs (Figure 2).
Relationship between the innate immune system and autoimmunity
As stated above, multiple studies have suggested a link between environmentally-induced inflammation and autoimmunity. Infectious agents can induce autoimmune diseases in both experimental and clinical settings [2]. Likewise, an association of an infectious disease with outbreaks of autoimmune-type diabetes mellitus has been reported. A variety of mechanisms have been invoked to explain these observations. One attractive hypothesis is that licensing of antigen-presenting cells by microbial stimuli may represent a checkpoint for functional self-tolerance and may provide a "third signal", mimicking costimulatory signaling [52, 53]. Therefore, microorganisms unrelated to the autoantigens that have primed the autoreactive T cells could dictate the onset and exacerbation of autoimmune diseases. Toll-like receptor (TLR) engagement by pathogen-associated molecular patterns (PAMPs) has been shown to be an important mechanism for optimal cellular immune responses [53]. APC TLR engagement indirectly enhances activated CD4+ T cell proliferation, differentiation, and survival by promoting the up-regulation of costimulatory molecules and the secretion of proinflammatory cytokines. TLR therefore seems to link activation of innate immunity to immune disorders. Innate immune cells, such as APC engulf pathogens by phagocytosis, and present pathogen-derived peptide antigens to naïve T cells. In addition, TLRs recognizing pathogen-derived components induce expression of genes, such as costimulatory molecules and inflammatory cytokines. Phagocytosis-mediated antigen presentation, together with TLR-mediated expression of costimulatory molecules and inflammatory cytokines, instruct development of antigen-specific adaptive immunity, especially Th1 cells, a T cell phenotype associated with T1DM [54]. Thus, as progress is made in the field, it will be important to combine costimulation blockade with inhibitors of innate immunity to maximize the therapeutic value of each.
Interventional trials: the use of T cell costimulatory pathways
In the absence of full costimulation, specific antigen recognition leads to partial T cell activation, T cell anergy and/or T cell deletion. This observation led the way for interventional strategies in autoimmune diseases and organ transplantation. Interventional trials in patients with rheumatoid arthritis have used costimulation blockade by a fusion protein-cytotoxic T-lymphocyte-associated antigen 4-IgG1 (CTLA4Ig) [55]. CTLA4Ig binds to CD80 and CD86 on antigen-presenting cells, blocking the engagement of CD28 on T cells and preventing T-cell activation (Figure 2). In patients with active rheumatoid arthritis, which is characterized by an ongoing T cell recruitment and activation, treatment with CTLA4Ig significantly improved signs and symptoms. These data suggest that costimulatory pathways may provide novel clues to immuno-pathology and treatment of ongoing islet cell autoimmunity. More recently, a second generation high affinity mutant form of CTLA4Ig (LEA2Y/belatacept) has been developed that binds CD80 twice as effectively as CTLA4Ig and CD86 four times more effectively. In large animal studies, the avidity of the new agent produced greater efficacy, setting the stage for long term costimulation blockade in the autoimmunity and transplant setting. Currently, this drug is in clinical trials in kidney transplantation and represents an important step forward in the development of CTLA4Ig therapies. This is a kind of artificial modulation of a system of molecules on the surface of T cells that has been designed to specifically provide on-off switches to support or abrogate the activation of T cells. This approach holds considerable potential because it avoids toxicities associated with cell lysis, while theoretically specifically affecting only those T cells which are being continuously stimulated to become activated. Interestingly, it has been suggested that one mechanism of action of this new therapy has been the induction of IDO in APCs as a consequence of CTLA-4 binding [56, 57] (Figure 2). Currently no results of trials in T1DM have been published using this approach [15].
Conclusion
Currently the importance of the various diabetes-associated genes is becoming clearer due to functional studies. Data have emerged in recent years, showing that the members of the B7 and CD28 families play a crucial role in T cell activities seen in various autoimmune disorders, including T1DM. The findings of these studies provide a classical example of functional genomics. The IDDM12 locus which is linked to chromosome 2q33 has revealed that the CTLA-4 gene is a prototype candidate gene in T1DM. Functional studies, including the use of transgenic or knockout animal models showed that CTLA-4 plays a major role in down-regulating T cell activation and maintaining immunologic homeostasis. Therefore costimulatory signaling is instrumental in controlling potentially autoreactive T cells that have escaped negative selection in the thymus. CTLA-4 ligation following T cell activation, down-regulates cytokine production and cell-cycle progression. CTLA-4 reveals a dynamic role at the immunological synapse leading to direct attenuation of early cell signals.
Overall the delineation of costimulatory pathways has broadened our understanding in the control of immune responses at the molecular level, and led to identification of molecular targets for potential treatment of autoimmune diseases. Since the B7-CD28 family molecules play a critical role in the control of initiation, progression and pathogenesis of autoimmune diseases, these molecules are a highly likely target. The understanding of a complex interplay within the immunological synapse with priming, activation, maturation and amplification of immune responses will therefore lead to a more rationale-based design of interventional strategies, thus leading to new methods and approaches for the management of autoimmune diseases. Some B7 homologues have unknown receptors, indicating that other immunoregulatory pathways remain to be described and will leave enough space for novel studies.
Is immunomodulation a valuable treatment option of T1DM in the prediabetic phase, and at onset only? Pancreas and insulin-producing β-cell transplantation will extend the scope of immunomodulation. Provided that the supply for insulin-producing β-cells has been resolved, most likely by stem cell technology, the ongoing modulation of autoreactive T cells will become a key issue in T1DM treatment. The studies currently available dealing with costimulatory signaling have already provided novel avenues to overcome existing hurdles for the promising approaches of cell-based therapy in T1DM. Especially the usage of immunoinhibitory receptor pathways might therefore be of considerable importance in the near future as a treatment target. One might speculate that the increased understanding of costimulatory pathways of T-cell regulation results in promising new therapeutics because T-cell interference can be better targeted to specific states of activation or location. This may decrease side-effects of standard immunosuppressive therapy such as systemic immunosuppression.
Acknowledgments:
This work is supported by grants from the German Research Council (DFG; SFB 518, DFG-Graduiertenkolleg "Molekulare Diabetologie und Endokrinologie"), and the NIH to BOB (NIH-DK 62418). JAB is supported by the JDRF and the NIAID, NIH.
References
- Jerne NK. Towards a network theory of the immune system. Annals d 'Imunnologie (Institute Pasteur) 1974. 125 C:373-389. [DOD]
- Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 2002. 347:911-920. [DOD] [CrossRef]
- Durinovic-Bello I, Schlosser M, Riedl M, Maisel N, Rosinger S, Kalbacher H, Deeg M, Ziegler M, Elliott J, Roep BO, Karges W, Boehm BO. Pro- and anti-inflammatory cytokine production by autoimmune T cells against preproinsulin in HLA-DRB1*04, DQ8 Type 1 diabetes. Diabetologia 2004. 47:439-450. [DOD] [CrossRef]
- Roep BO, Atkinson MA, Van Endert PM, Gottlieb PA, Wilson SB, Sachs JA. Autoreactive T cell responses in insulin-dependent (type 1) diabetes mellitus. Report of the first international workshop for standardization of T cell assays. J Autoimmun 1999. 13:267-282. [DOD] [CrossRef]
- June CH, Ledbetter JA, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today 1990. 11:211-216. [DOD]
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998. 18(5):389-418. [DOD]
- Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest 2004. 114(7):979-987. [DOD] [CrossRef]
- Noel PJ, Boise LH, Thompson CB. Regulation of T cell activation by CD28 and CTLA4. Adv Exp Med Biol 1996. 406:209-217. [DOD]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science 1999. 285:221-227. [DOD] [CrossRef]
- Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol 2003. 3:973-983. [DOD]
- Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol 2002. 2:116-126. [DOD] [CrossRef]
- Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin HY, Fuchs E, Singh B, Thompson CB, Bluestone JA. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity 1996. 5(3):285-293. [DOD] [CrossRef]
- Nakae S, Asano M, Horai R, Sakaguchi N, Iwakura Y. IL-1 enhances T cell-dependent antibody production through induction of CD40 ligand and OX40 on T cells. J Immunol 2001. 167(1):90-97. [DOD]
- Durie FH, Foy TM, Masters SR, Laman JD, Noelle RJ. The role of CD40 in the regulation of humoral and cell-mediated immunity. Immunol Today 1994. 15(9):406-411. [DOD] [CrossRef]
- Anand S, Chen L. Control of autoimmune diseases by the B7-CD28 family molecules. Curr Pharm Des 2004. 10:121-128. [DOD] [CrossRef]
- McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, Chernova T, Malenkovich N, Jabs C, Kuchroo VK, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol 2000. 165(9):5035-5040. [DOD]
- Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity 1999. 11(4):423-432. [DOD] [CrossRef]
- Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, Linsley PS. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med 1994. 180(6):2049-2058. [DOD] [CrossRef]
- Fallarino F, Fields PE, Gajewski TF. B7-1 engagement of cytotoxic T-lymphocyte antigen 4 inhibits T cell activation in the absence of CD28. J Exp Med 1998. 188(1):205-210. [DOD] [CrossRef]
- Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB, Bluestone JA. Molecular basis of T cell inactivation by CTLA-4. Science 1998. 282(5397):2263-2266. [DOD] [CrossRef]
- Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, Gardner JP, Hambor JE, Neveu MJ, Thompson CB. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 2000. 13(3):313-322. [DOD] [CrossRef]
- Baroja ML, Vijayakrishnan L, Bettelli E, Darlington PJ, Chau TA, Ling V, Collins M, Carreno BM, Madrenas J, Kuchroo VK. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J Immunol 2002. 168(10):5070-5078. [DOD]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000. 192(7):1027-1034. [DOD] [CrossRef]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001. 2(3):261-268. [DOD] [CrossRef]
- Khoury SJ, Sayegh MH. The roles of the new negative T cell costimulatory pathways in regulating autoimmunity. Immunity 2004. 20:529-538. [DOD] [CrossRef]
- Khattri R, Auger JA, Griffin MD, Sharpe AH, Bluestone JA. Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J Immunol 1999. 162(10):5784-5791. [DOD]
- Mandelbrot DA, McAdam AJ, Sharpe AH. B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). J Exp Med 1999. 189:435-440. [DOD] [CrossRef]
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995. 3:541-547. [DOD] [CrossRef]
- Watts TH, DeBenedette MA. T cell co-stimulatory molecules other than CD28. Curr Opin Immunol 1999. 11:286-293. [DOD] [CrossRef]
- Pociot F, McDermott MF. Genetics of type 1 diabetes mellitus. Genes Immun 2002. 3:235-349. [DOD] [CrossRef]
- Redondo MJ, Eisenbarth GS. Genetic control of autoimmunity in type 1 diabetes and associated disorders. Diabetologia 2002. 45:605-622. [DOD] [CrossRef]
- Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003. 423:506-511. [DOD] [CrossRef]
- Marron MP, Zeidler A, Raffel LJ, Eckenrode SE, Yang JJ, Hopkins DI, Garchon HJ, Jacob CO, Serrano-Rios M, Martinez Larrad MT, et al. Genetic and physical mapping of a type 1 diabetes susceptibility gene (IDDM12) to a 100-kb phagemid artificial chromosome clone containing D2S72-CTLA4-D2S105 on chromosome 2q33. Diabetes 2000. 49:492-499. [DOD]
- Mein CA, Esposito L, Dunn MG, Johnson GCL, Timms AE, Goy JV, Smith AN, Sebag-Montefiore L, Merriman ME, Wilson AJ, et al. A search for type 1 diabetes susceptibility genes in families from the United Kingdom. Nat Genet 1998. 19:297-300. [DOD] [CrossRef]
- Kouki T, Sawai Y, Gardine CA, Fisfalen ME, Alegre ML, DeGroot LJ. CTLA-4 gene polymorphism at position 49 in exon 1 reduces the inhibitory function of CTLA-4 and contributes to the pathogenesis of Graves' disease. J Immunol 2000. 165:6606-6611. [DOD]
- Vijayakrishnan L, Slavik JM, Illes Z, Greenwald RJ, Rainbow D, Greve B, Peterson LB, Hafler DA, Freeman GJ, Sharpe AH, Wicker LS, Kuchroo VK. An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity 2004. 20:563-575. [DOD] [CrossRef]
- Magistrelli G, Jeannin P, Herbault N, Benoit De Coignac A, Gauchat JF, Bonnefoy JY, Delneste Y. A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur J Immunol 1999. 29(11):3596-3602. [DOD] [CrossRef]
- Wicker LS, Chamberlain G, Hunter K, Rainbow D, Howlett S, Tiffen P, Clark J, Gonzalez-Munoz A, Cumiskey AM, Rosa RL, et al. Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J Immunol 2004. 173(1):164-173. [DOD]
- Schwartz JC, Zhang X, Nathenson SG, Almo SC. Structural mechanisms of costimulation. Nat Immunol 2002. 3:427-434. [DOD] [CrossRef]
- Pechhold K, Patterson NB, Blum C, Fleischacker CL, Boehm BO, Harlan DM. Low dose streptozotocin-induced diabetes in rat insulin promoter-mCD80-transgenic mice is T cell autoantigen-specific and CD28-dependent. J Immunol 2001. 166(4):2531-2539. [DOD]
- Pechhold K, Karges W, Blum C, Boehm BO, Harlan DM. Βeta-cell-specific CD80 (B7-1) expression disrupts tissue protection from autoantigen-specific CTL-mediated diabetes. J Autoimmun 2003. 20(1):1-13. [DOD] [CrossRef]
- Havari E, Lennon-Dumenil AM, Klein L, Neely D, Taylor JA, McInerney MF, Wucherpfennig KW, Lipes MA. Expression of the B7.1 costimulatory molecule on pancreatic beta-cells abrogates the requirement for CD4 T cells in the development of type 1 diabetes. J Immunol 2004. 173:787-796. [DOD]
- Karges W, Pechhold K, Al Dahouk S, Riegger I, Rief M, Wissmann A, Schirmbeck R, Barth C, Boehm BO. Induction of autoimmune diabetes through insulin (but not GAD65) DNA vaccination in nonobese diabetic and in RIP-B7.1 mice. Diabetes 2002. 51(11):3237-3244. [DOD]
- Lenschow DJ, Ho SC, Sattar H, Rhee L, Gray G, Nabavi N, Herold KC, Bluestone JA. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med 1995. 181(3):1145-1155. [DOD] [CrossRef]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000. 12(4):431-440. [DOD] [CrossRef]
- Rulifson IC, Sperling AI, Fields PE, Fitch FW, Bluestone JA. CD28 costimulation promotes the production of Th2 cytokines. J Immunol 1997. 158(2):658-665. [DOD]
- Bour-Jordan H, Grogan JL, Tang Q, Auger JA, Locksley RM, Bluestone JA. CTLA-4 regulates the requirement for cytokine-induced signals in T(H)2 lineage commitment. Nat Immunol 2003. 4(2):182-188. [DOD] [CrossRef]
- Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol 2001. 19:225-252. [DOD] [CrossRef]
- Eggena MP, Walker LS, Nagabhushanam V, Barron L, Chodos A, Abbas AK. Cooperative roles of CTLA-4 and regulatory T cells in tolerance to an islet cell antigen. J Exp Med 2004. 199:1725-1730. [DOD]
- Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T-lymphocyte-associated antigen 4. J Exp Med 2000. 192(2):303-310. [DOD] [CrossRef]
- Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol 2004. 34(11):2996-3005. [DOD] [CrossRef]
- Darabi K, Karulin AY, Boehm BO, Hofstetter HH, Fabry Z, LaManna JC, Chavez JC, Tary-Lehmann M, Lehmann PV. The third signal in T cell-mediated autoimmune disease? J Immunol 2004. 173:92-99. [DOD]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 2005. 17:1-14. [DOD] [CrossRef]
- Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, Odermatt B, Conrad C, Ittner LM, Bauer S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med 2005. 11:138-145. [DOD] [CrossRef]
- Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker JC, Hagerty DT, Moreland LW. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med 2003. 349(20):1907-1915. [DOD] [CrossRef]
- Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol 2002. 3(11):1097-1101. [DOD] [CrossRef]
- Boasso A, Herbeuval JP, Hardy AW, Winkler C, Shearer GM. Regulation of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA-synthetase by CTLA-4-Fc in human CD4+ T cells. Blood 2004. online pre-publishing October 2004. [DOD]
This article has been cited by other articles:

|
A proinsulin74-90-derived protease-resistant, altered peptide ligand increases TGF-beta1 secretion in PBMC from patients with type 1 diabetes mellitus
van Aalst D, Kalbacher H, Palesch D, Zou F, Spyrantis A, Rosinger S, Boehm BO, Burster T
J Leukoc Biol 2010. 87(5):943-948
|
|

|
Protease-resistant human GAD-derived altered peptide ligands decrease TNF-alpha and IL-17 production in peripheral blood cells from patients with type 1 diabetes mellitus
Boehm BO, Rosinger S, Sauer G, Manfras BJ, Palesch D, Schiekofer S, Kalbacher H, Burster T
Mol Immunol 2009. 46(13):2576-2584
|
|

|
Coexpression of CD25 and OX40 (CD134) receptors delineates autoreactive T-cells in type 1 diabetes
Endl J, Rosinger S, Schwarz B, Friedrich SO, Rothe G, Karges W, Schlosser M, Eiermann T, Schendel DJ, Boehm BO
Diabetes 2006. 55(1):50-60
|
|

|
Genetic Susceptibility to Type 1 Diabetes in the Intracellular Pathway of Antigen Processing – A Subject Review and Cross-Study Comparison
Sia C, Weinem M
Rev Diabet Stud 2005. 2(1):40-52
|
|

|
The Role of Regulatory T Cell Defects in Type I Diabetes and the Potential of these Cells for Therapy
Thomas D, Zaccone P, Cooke A
Rev Diabet Stud 2005. 2(1):9-18
|
|
|


)
)

