Review
| Rev Diabet Stud,
2004,
1(4):165-174 |
DOI 10.1900/RDS.2004.1.165 |
Involvement of Oxidative Stress and the JNK Pathway in Glucose Toxicity
Hideaki Kaneto, Yoshihisa Nakatani, Dan Kawamori, Takeshi Miyatsuka, Taka-aki Matsuoka
Department of Internal Medicine and Therapeutics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Address correspondence to: Hideaki Kaneto, e-mail: kaneto@medone.med.osaka-u.ac.jp
Keywords: diabetes, insulin resistance, insulin biosynthesis, oxidative stress, JNK pathway
Abstract
The hallmark of type 2 diabetes is pancreatic β-cell dysfunction and insulin resistance. Normal β-cells can compensate for insulin resistance by increasing insulin secretion, but insufficient compensation leads to the onset of glucose intolerance. Once hyperglycemia becomes apparent, β-cell function gradually deteriorates and insulin resistance becomes aggravated. Such phenomena are collectively called "glucose toxicity". Under diabetic conditions, oxidative stress is induced and the JNK pathway is activated, which is involved in "glucose toxicity". Activation of the JNK pathway suppresses insulin biosynthesis and interferes with insulin action. Indeed, suppression of the JNK pathway in diabetic mice improves insulin resistance and ameliorates glucose tolerance. Consequently, the JNK pathway plays a crucial role in the progression of pancreatic β-cell dysfunction and insulin resistance and thus could be a potential therapeutic target for the "glucose toxicity" found in diabetes.
Oxidative stress is induced under hyperglycemic conditions
The development of type 2 diabetes is usually associated with a combination of pancreatic β-cell dysfunction and insulin resistance. Normal β-cells can compensate for insulin resistance by increasing insulin secretion or β-cell mass, but insufficient compensation leads to the onset of glucose intolerance. Chronic hyperglycemia is a cause of impaired insulin biosynthesis and secretion; once hyperglycemia becomes apparent, β-cell function gradually deteriorates and insulin resistance becomes aggravated [1-5]. This process is called "glucose toxicity".
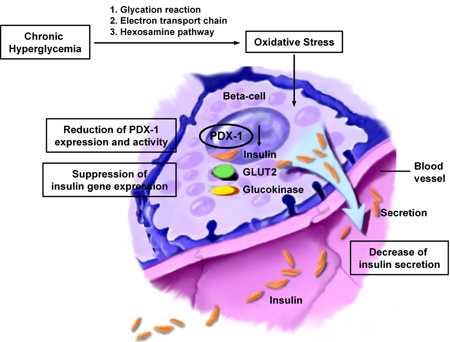
Under diabetic conditions, reactive oxygen species (ROS) increase in various tissues and are involved in the development of diabetic complications [6-9]. Pancreatic β-cells have recently emerged as a target of oxidative stress-mediated tissue damage [10-25]. β-cells express abundant levels of the high-Km glucose transporter GLUT2 and thereby display highly efficient glucose uptake when exposed to high glucose concentration. Accordingly, extracellular hyperglycemia causes intracellular hyperglycemia in β-cells, leading to the induction of ROS in pancreatic islets of diabetic animals. Indeed, it was shown that the presence of oxidative stress markers 8-hydroxy-2'-deoxyguanosine (8-OHdG) and 4-hydroxy-2,3-nonenal (4-HNE) is increased in islets under diabetic conditions [13, 21]. In addition, due to the relatively low expression of antioxidant enzymes such as catalase, and glutathione peroxidase [12], β-cells are rather vulnerable to oxidative stress. Thus, it is likely that oxidative stress is involved in β-cell deterioration in type 2 diabetes. There are several sources of ROS production in cells: 1. The non-enzymatic glycosylation reaction [10, 11], 2. the electron transport chain in mitochondria [8, 22], and 3. the hexosamine pathway [19] (Figure 1). In diabetic animals, glycation can be observed in various tissues and organs, and various kinds of glycated proteins are produced in a non-enzymatical manner through the Maillard reaction. During the reaction which in turn produces Schiff base, Amadori product and advanced glycosylation end products (AGE), ROS are also produced [10]. Also, the electron transport chain in mitochondria is likely to be an important pathway triggering the production of ROS. Indeed, it has been suggested that mitochondrial overwork, which causes the emergence of ROS, is a potential mechanism causing impaired first-phase glucose-stimulated insulin secretion found in the early stage of diabetes [22] as well as diabetic complications [8, 9].
 |
 |
Figure 1. Oxidative stress and pancreatic β-cell dysfunction. Extra-cellular hyperglycemia contributes to intra-cellular hyper-glycemia via the following ways of ROS production: 1. The non-enzymatic glycosylation reaction, 2. the electron transport chain in mitochondria, and 3. the hexosamine pathway. DNA-binding activity of PDX-1 is rather sensitive to oxidative stress. ROS production in pancreatic β-cells, therefore, leads to a decreased expression of PDX-1, which in turn causes the down-regulation of β-cell genes, such as insulin, GLUT2, and glucokinase. Thus, chronic hyperglycemia suppresses insulin biosynthesis and secretion. |
|
Oxidative stress is involved in pancreatic β-cell dysfunction
The glycation reaction suppresses the insulin gene transcription in β-cells by provoking oxidative stress. When β-cell-derived HIT cells were exposed to D-ribose, a strong reducing sugar, insulin gene promoter activity and mRNA expression were suppressed, whereas no such changes were observed with the β-actin gene [11]. These were neutralized with aminoguanidine, an inhibitor of the glycation reaction, or N-acetyl-L-cysteine, an antioxidant, indicating that D-ribose suppresses insulin gene transcription by provoking oxidative stress through the glycation reaction.
Pancreatic and duodenal homeobox factor-1 (PDX-1), also known as IDX-1/STF-1/IPF1 [26-28], is a member of the homeodomain-containing transcription factor family. PDX-1 is expressed in the pancreas and duodenum and plays a crucial role in pancreas development [29-38], β-cell differentiation/regeneration [39-51], and in maintaining normal β-cell function by regulating many important β-cell genes, including insulin, GLUT2, and glucokinase [52-62]. During the development of the pancreas, PDX-1 expression is maintained in multipotential precursors that co-express several hormones and later it becomes restricted to β-cells. There is no pancreas in mice homozygous for a targeted mutation in the PDX-1 gene and the mice develop fatal perinatal hyperglycemia [29]. Heterozygous PDX-1-deficient mice reveal impaired glucose tolerance [63], which also provides evidence for the crucial role of PDX-1 in pancreas development. Clinically, mutations in PDX-1 are known to cause some cases of maturity-onset diabetes of the young (MODY) [64]. As a possible cause of the reduction in the insulin gene promoter activity by oxidative stress, we found that the DNA-binding activity of PDX-1 is rather sensitive to oxidative stress; when HIT cells were exposed to D-ribose, PDX-1 binding to the insulin gene was markedly reduced, which was prevented by aminoguanidine or NAC [11]. In summary, chronic hyperglycemia suppresses insulin biosynthesis and secretion by provoking oxidative stress through various pathways, accompanied by the reduction of PDX-1 DNA-binding activity (Figure 1).
Because oxidative stress is produced under diabetic conditions and is likely to be involved in pancreatic β-cell dysfunction found in diabetes, we evaluated the potential usefulness of antioxidants in treatment for type 2 diabetes. We treated obese diabetic C57BL/KsJ-db/db mice with antioxidants (N-acetyl-L-cysteine plus vitamin C and E) and evaluated their effects [14]. The antioxidant treatment retained glucose-stimulated insulin secretion and moderately decreased blood glucose levels. The β-cell mass was significantly larger in the mice treated with the antioxidants. The antioxidant treatment suppressed apoptosis in β-cells without changing the rate of β-cell proliferation. The amounts of insulin content and insulin mRNA were also retained by the antioxidant treatment. Furthermore, PDX-1 expression was more clearly visible in the nuclei of islet cells after the antioxidant treatment [14]. Similar effects were observed with the Zucker diabetic fatty (ZDF) rat, another animal model for type 2 diabetes [15]. In summary, these data indicate that antioxidant treatment can generate beneficial effects for diabetes with preservation of in vivo β-cell function.
In addition, as a step toward the clinical trial of antioxidants for type 2 diabetes, we investigated the possible anti-diabetic effects of probucol, an antioxidant widely used as an anti-hyperlipidemic agent, on the preservation of β-cell function in diabetic C57BL/KsJ-db/db mice [21]. Immunostaining for oxidative stress markers such as 4-hydroxy-2-nonenal (HNE)-modified proteins and heme oxygenase-1 revealed that probucol treatment decreased reactive oxygen species (ROS) in pancreatic islets of diabetic animals. Probucol treatment preserved β-cell mass, the insulin content, and glucose-stimulated insulin secretion, leading to the improvement of glucose tolerance [21]. These data suggest the potential usefulness of antioxidants for treating diabetes and provide further evidence for the implication of oxidative stress in β-cell glucose toxicity found in diabetes (Figure 1).
The JNK pathway is involved in pancreatic β-cell dysfunction
Several signal transduction pathways including c-Jun N-terminal kinase (JNK) (also known as stress-activated protein kinase (SAPK)) [65-68], p38 mitogen-activated protein kinase (p38 MAPK), and protein kinase C (PKC) are activated by oxidative stress in several cell types including pancreatic β-cells. It has been shown recently that activation of JNK is involved in the reduction of insulin gene expression by oxidative stress and that suppression of the JNK pathway can protect β-cells from oxidative stress [69]. When isolated rat islets were exposed to oxidative stress, JNK, p38 MAPK, and PKC pathways were activated, preceding the decrease of insulin gene expression. Adenovirus-mediated over-expression of dominant-negative type (DN) JNK, but not the p38 MAPK inhibitor SB203580 nor the PKC inhibitor GF109203X, protected insulin gene expression and secretion from oxidative stress. In contrast, wild type (WT) JNK over-expression suppressed both insulin gene expression and secretion.
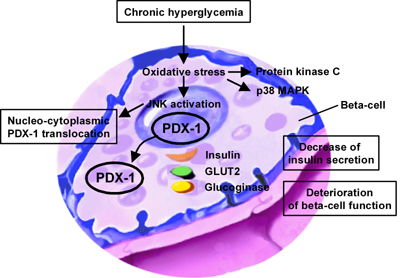
These results are correlated with changes in the binding of the major transcription factor PDX-1 to the insulin promoter; adenoviral over-expression of DN-JNK preserved PDX-1 DNA-binding activity in the presence of oxidative stress, while WT-JNK over-expression decreased PDX-1 DNA-binding activity. PDX-1 DNA-binding activity is decreased in association with a reduction of insulin gene transcription after chronic exposure to high glucose concentration. The impairment of PDX-1 activity and subsequent suppression of insulin gene transcription is mediated by the activation of the JNK pathway in the diabetic state. Thus, it is likely that JNK-mediated suppression of PDX-1 DNA-binding activity accounts to some extent for the deterioration of the β-cell function (Figure 2).
 |
|
Figure 2. The JNK pathway and pancreatic β-cell dysfunction. Signal transduction pathways including JNK, p38 mitogen-activated protein kinase (p38 MAPK), and protein kinase C (PKC) are activated by oxidative stress in pancreatic β-cells. Wild-type-JNK over-expression decreases PDX-1 DNA-binding activity in association with a reduction of insulin gene transcription after chronic exposure to high glucose concentration leading to a decrease of insulin secretion. In addition, PDX-1 is translocated pathogenically from the nucleus to the cytoplasm by JNK activation. |
|
As a potential mechanism for JNK-mediated PDX-1 inactivation, we recently reported that PDX-1 is translocated from the nuclei to the cytoplasm in response to oxidative stress; when oxidative stress was charged upon β-cell-derived HIT cells, both intrinsically expressed PDX-1 and exogenously introduced green fluorescent protein (GFP)-tagged PDX-1 moved from the nuclei to the cytoplasm [70] (Figure 2). Addition of DN-JNK inhibited the oxidative stress-induced PDX-1 translocation, suggesting an essential role of JNK in mediating the phenomenon. Whereas the nuclear localization signal (NLS) in PDX-1 was not affected by oxidative stress, leptomycin B, a specific inhibitor of the classical, leucine-rich nuclear export signal (NES), inhibited nucleo-cytoplasmic translocation of PDX-1 induced by oxidative stress. Indeed, we identified an NES at position 82-94 of the mouse PDX-1 protein (Figure 2) [70].
Toexamine whether DN-JNK can protect β-cells from the toxic effects of hyperglycemia and to explore the potential therapeutic application for islet transplantation, we performed islet transplantation into diabetic mice [69]. Isolated rat islets were infected with dominant-negative JNK expressing adenovirus (Ad-DN-JNK) or Ad-GFP and cultured for 2 days; then 500 islets were transplanted under kidney capsules of STZ-induced diabetic Swiss nude mice. Blood glucose levels were not sufficiently decreased by the transplantation of islets infected with Ad-GFP, which was probably due to toxic effects of hyperglycemia upon a marginal islet number, but were markedly decreased by Ad-DN-JNK. Four weeks after the transplantation of islets infected with Ad-GFP, insulin mRNA levels in islet grafts were clearly decreased compared with those before transplantation, but relatively preserved by DN-JNK overexpression [69]. These results suggest that DN-JNK can protect β-cells from some of the toxic effects of hyperglycemia during this transplant period, providing new insights into the mechanism through which oxidative stress suppresses insulin gene transcription in β-cells. Also, the finding that this adverse outcome can be prevented by DN-JNK over-expression suggests that the JNK pathway in β-cells could become a new therapeutic target for diabetes.
It is known that β-cell destruction by cytokines such as interleukin-1β (IL-1β) [71-74] can be prevented by inhibition of the JNK pathway [75-78], implying that JNK plays a role in autoimmune β-cell destruction found in early stages of type 1 diabetes. Also, it has been reported that levels of 8-hydroxy-2'-deoxy-guanosine (8-OHdG), a marker for oxidative stress, are increased in the blood of type 2 diabetic patients as well as in islets of type 2 diabetic animal models [6, 13, 21] and that JNK activation by oxidative stress in islets actually reduces the PDX-1 DNA binding activity and insulin gene transcription [69]. In addition, the significance of JNK in the development of diabetes comes from the result of a genetic analysis in humans; while islet-brain-1 (IB1), the human and rat homologue of mouse JNK-interacting protein-1 (JIP-1) [79, 80], was known to selectively inhibit the JNK signaling [78], it was reported that a missense mutation within the IB1-encoding MAPKIP1 gene (S59N) is associated with a late onset type 2 diabetes [81]. Thus, we assume that JNK is involved in the deterioration of β-cell function in both type 2 diabetes and the early stage of type 1 diabetes.
The JNK pathway is involved in insulin resistance
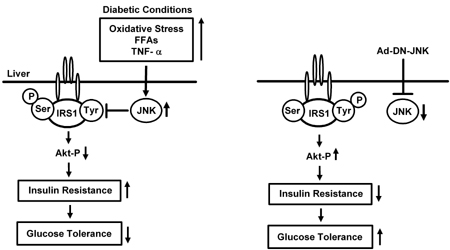
The JNK pathway is known to be activated under diabetic conditions and possibly to be involved in the progression of insulin resistance. We have recently examined the effects of a modulation of the JNK pathway in the liver on insulin resistance and glucose tolerance [82]. Overexpression of dominant-negative type JNK in the liver of obese diabetic mice dramatically improved insulin resistance and markedly decreased blood glucose levels. When C57BL/KsJ-db/db mice were treated with Ad-DN-JNK, non-fasting blood glucose levels were markedly reduced, whereas no such effect was observed in Ad-GFP-treated mice. In the intraperitoneal insulin tolerance test (IPITT), the hypoglycemic response to insulin was larger in Ad-DN-JNK-treated C57BL/KsJ-db/db mice compared to Ad-GFP-treated mice. To investigate this point further, we performed the euglycemic hyperinsulinemic clamp test. Glucose infusion rate (GIR) in Ad-DN-JNK-treated mice was higher than that in Ad-GFP-treated mice, indicating that suppression of the JNK pathway in the liver reduces insulin resistance and thus ameliorates glucose tolerance in C57BL/KsJ-db/db mice. Furthermore, hepatic glucose production (HGP) was significantly lower in Ad-DN-JNK-treated mice. In contrast, there was no difference in the glucose disappearance rate (Rd) between these two groups. These results indicate that the reduction of insulin resistance and the amelioration of glucose tolerance by DN-JNK overexpression are mainly effectuated by a suppression of hepatic glucose production (Figure 3).
 |
|
Figure 3. The effects of the JNK pathway on insulin resistance and glucose tolerance. The JNK pathway in the liver is activated by factors such as oxidative stress, free fatty acids (FFAs), and TNF-α. The levels of these factors are elevated under obese diabetic conditions. Serine phosphorylation of insulin receptor substrate-1 (IRS-1) inhibits insulin-stimulated tyrosine phosphorylation of IRS-1 and leads to an increase in insulin resistance. Expression of wild type JNK in the liver of normal mice decreased insulin sensitivity. Suppression of the JNK pathway by the treatment with Ad-DN-JNK enhances insulin signaling which leads to a decrease in gluconeogenesis and amelioration of glucose tolerance. |
|
It has been reported that serine phosphorylation of insulin receptor substrate-1 (IRS-1) inhibits insulin-stimulated tyrosine phosphorylation of IRS-1, leading to an increase in insulin resistance [83, 84]. IRS-1 serine 307 phosphorylation was markedly decreased in Ad-DN-JNK-treated mice. We also found an increase in IRS-1 tyrosine phosphorylation in Ad-DN-JNK-treated mice compared to control mice. Reduction of Akt serine 473 phosphorylation was observed in Ad-DN-JNK-treated C57BL/KsJ-db/db mice. Therefore, an increase in IRS-1 serine phosphorylation may be closely associated with the development of insulin resistance induced by JNK overexpression. Next, we examined the expression levels of the key gluconeogenic enzymes, phosphoenolpyruvate carboxykinase (PEP CK) and glucose-6-phosphatase (G6Pase), both of which are known to be regulated by insulin signaling. Expression levels of both enzymes were markedly decreased by Ad-DN-JNK treatment in C57BL/KsJ-db/db mice. These results indicate that suppression of the JNK pathway enhances insulin signaling which leads to a decrease in gluconeogenesis and amelioration of glucose tolerance. Similar effects were observed in high-fat/high-sucrose diet-induced diabetic mice. Conversely, expression of wild type JNK in the liver of normal mice decreased insulin sensitivity. In summary, these findings suggest that suppression of the JNK pathway in liver exerts greatly beneficial effects on insulin resistance status and glucose tolerance in both genetic and dietary models of diabetes (Figure 3).
The JNK pathway is known to be activated by several factors such as oxidative stress, free fatty acids (FFAs), tumor necrosis factor-α (TNF-α), all of which are known to be increased under diabetic conditions. Under diabetic conditions, reactive oxygen species (ROS) are produced in various tissues and are involved in the development of insulin resistance as well as the progression of β-cell deterioration. FFAs and TNF-α are also likely to be involved in the development of insulin resistance; levels of FFAs and TNF-α are increased under obese diabetic conditions with insulin resistance, which leads to a further increase in insulin resistance. Thus, we assume that the improvement of insulin resistance by suppression of the JNK pathway is, at least in part, counterbalancing the deleterious effects of several factors such as oxidative stress, FFAs and TNF-α (Figure 3).
It has been also reported recently that JNK activity is abnormally elevated in liver, muscle and adipose tissues in obese type 2 diabetic mice and that insulin resistance is substantially reduced in mice homozygous for a targeted mutation in the JNK1 gene (JNK-KO mice) [83]. When JNK-KO mice are placed on a high-fat/high-caloric nutrition, obese wild type mice develop mild hyperglycemia compared to lean wild-type control mice. In contrast, blood glucose levels in obese JNK-KO mice were proven to be significantly lower than those in obese wild type mice. In addition, serum insulin levels in obese JNK-KO mice were significantly lower than those in obese wild type mice. Intraperitoneal insulin tolerance tests showed that hypoglycemic response to insulin in obese wild type mice is lower than that in obese JNK-KO mice. Also, the intraperitoneal glucose tolerance test revealed a higher degree of hyperglycemia in obese wild type mice than in obese JNK-KO mice. These results indicate that the JNK-KO mice are protected from the development of dietary obesity-induced insulin resistance. Furthermore, targeted mutations in JNK were introduced in genetically obese mice (ob/ob) [65]. Blood glucose levels in ob/ob-JNK-KO mice were lower than those in ob/ob wild type mice, and the ob/ob wild type mice displayed a severe and progressive hyperinsulinemia. Thus, JNK deficiency can provide partial resistance against obesity, hyperglycemia and hyperinsulinemia in both genetic and dietary models of diabetes.
Consequently, obese type 2 diabetes is associated with activation of the JNK pathway, and the absence of JNK results in substantial protection from obesity-induced insulin resistance. These results strongly suggest that JNK plays a crucial role in the progression of insulin resistance found in type 2 diabetes. Therefore, a selective interference with JNK activity is a potential therapeutic target for obesity, insulin resistance and type 2 diabetes [85, 86].
The JNK pathway as a potential therapeutic target for diabetes
Protein transduction domains (PTDs) such as the small PTD from the TAT protein of human immunodeficiency virus (HIV-1), the VP22 protein of Herpes simplex virus, and the third α-helix of the homeodomain of Antennapedia, a Drosophila transcription factor, are known to allow various proteins and peptides to be efficiently delivered into cells through the plasma membrane, and thus there has been increasing interest in their potential usefulness for the delivery of bioactive proteins and peptides into cells [87-93].
We have recently evaluated the potential usefulness of a JNK inhibitory peptide in the treatment of type 2 diabetes and found that the cell-permeable JNK inhibitory peptide (amino acid sequence: GRKKRRQRRRPPRPKRPTTLNLFPQVPRSQDT) is very effective. This peptide is derived from the JNK binding domain of JNK-interacting protein-1 (JIP-1), also known as islet-brain-1 (IB-1), and has been reported to function as a dominant inhibitor of the JNK pathway [76]. To convert the minimal JNK-binding domain into a bioactive cell-permeable compound, a 20-amino acid sequence derived from the JNK-binding domain of JIP-1 (RPK RPT TLN LFP QVP RSQ DT) was covalently linked to a 10-amino acid carrier peptide derived from the HIV-TAT sequence (GRK KRR QRR R); then to monitor peptide delivery, this JIP-1-HIV-TAT peptide was further conjugated with fluorescein isothiocyanate (FITC). First, to examine the effectiveness of the JNK inhibitory peptide in vivo, C57BL/KsJ-db/db obese diabetic mice were injected intraperitoneally with the JIP-1-HIV-TAT-FITC peptide. The FITC-conjugated peptide showed fluorescence signals in insulin target organs (liver, fat, muscle) and in insulin secreting tissue (pancreatic islets). Next, we examined whether the JNK pathway is inhibited after treatment with JIP-1-HIV-TAT-FITC. In various tissues (liver, fat, and muscle), JNK activity was actually suppressed by JIP-1-HIV-TAT-FITC in a dose-dependent manner.
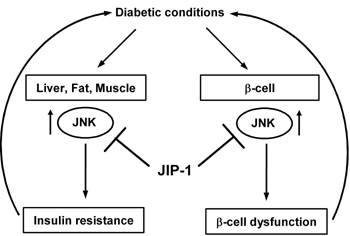
To investigate whether suppression of the JNK pathway exerts beneficial effects on diabetes, we treated C57BL/KsJ-db/db mice with an intraperitoneal injection of the JNK inhibitory peptide, JIP-1-HIV-TAT-FITC or a scramble peptide as a control. There was no difference in body weight and food intake between the JIP-1-HIV-TAT-FITC-treated and untreated mice. The non-fasting blood glucose levels in mice treated with JIP-1-HIV-TAT-FITC were significantly decreased compared to untreated mice or those treated with the scramble peptide. Also, the glucose tolerance test showed that glucose tolerance in JIP-1-HIV-TAT-FITC-treated mice was significantly ameliorated in comparison to untreated or scramble peptide-treated mice [94]. These data indicate that the JNK pathway is involved in the exacerbation of diabetes and that suppression of the JNK pathway could be a therapeutic target for diabetes (Figure 4).
 |
|
Figure 4. The JNK pathway as a therapeutic target for diabetes. Oxidative stress mediated by chronic diabetic conditions activates JNK in several cells, such as liver-, fat-, muscle-, as well as β-cells, leads to insulin resistance and β-cell dysfunction. The JNK pathway can be suppressed by JNK inhibitory peptide, JIP-1-HIV-TAT-FITC. Treatment with JIP-1 decreases non-fasting blood glucose levels and improves glucose tolerance in mice significantly. |
|
To investigate the possible effects of the JNK inhibitory peptide on insulin action, we performed an insulin tolerance test. Reduction of blood glucose levels in response to injected insulin was much larger in JIP-HIV-TAT-FITC-treated mice than in untreated mice, indicating that the peptide treatment improves insulin sensitivity. In contrast, no such effect was observed when non-diabetic C57BL6 mice were treated with JIP-1-HIV-TAT-FITC. These results imply that the JNK pathway is activated under diabetic conditions and thus the JNK inhibitory peptide exerts beneficial effects on insulin action and glucose tolerance.
To further investigate the effect of the peptide on insulin resistance, we performed the euglycemic hyperinsulinemic clamp test. The steady-state glucose infusion rate (GIR) in JIP-1-HIV-TAT-FITC-treated mice was significantly higher than in untreated mice, indicating that JIP-1-HIV-TAT-FITC reduces insulin resistance in C57BL/KsJ-db/db mice. Furthermore, we evaluated endogenous hepatic glucose production (HGP) and glucose disappearance rate (Rd) in the JNK inhibitory peptide-treated mice. It is noted that Rd reflects glucose utilization in the peripheral tissues. HGP in JIP-1-HIV-TAT-FITC-treated mice was significantly lower than in untreated mice. In addition, Rd in JIP-1-HIV-TAT-FITC-treated mice was significantly higher than in untreated mice. These results indicate that JIP-1-HIV-TAT-FITC treatment reduces insulin resistance by decreasing HGP and increasing Rd. The data provide strong evidence that JNK is indeed a crucial component of the biochemical pathway responsible for insulin resistance in vivo. In addition, to examine the effect of JIP-1-HIV-TAT-FITC treatment on insulin biosynthesis, we measured the insulin mRNA level and content in pancreata of C57BL/KsJ-db/db mice which had been treated with the peptide. The insulin mRNA level and insulin content were significantly higher in the peptide-treated mice. Thus, we assume that the JNK inhibitory peptide exerted some beneficial effects on the pancreatic islets.
To explore the molecular mechanism of how JIP-1-HIV-TAT-FITC treatment improves insulin sensitivity and ameliorates glucose tolerance, we evaluated IRS-1 serine 307 phosphorylation in various insulin target tissues (liver, fat, and muscle) of JIP-1-HIV-TAT-FITC-treated mice. IRS-1 serine 307 phosphorylation was decreased in JIP-1-HIV-TAT-FITC-treated mice compared to control mice. We also found an increase of IRS-1 tyrosine phosphorylation in the peptide-treated mice compared to control mice. Concomitantly, an increase of Akt serine 473 and threonine 308 phosphorylation, both of which are known to be important for activation of the Akt pathway, was observed in JIP-1-HIV-TAT-FITC-treated mice.
In conclusion, the cell-permeable JNK inhibitory peptide, JIP-1-HIV-TAT-FITC, improves insulin resistance and ameliorates glucose intolerance, indicating the critical involvement of the JNK pathway in diabetes and the usefulness of the cell-permeable JNK inhibitory peptide as a novel therapeutic agent for diabetes (Figure 4).
Conclusions
Activation of the JNK pathway is involved in the progression of insulin resistance as well as deterioration of pancreatic β-cell function. Indeed, suppression of the JNK pathway in obese diabetic mice markedly improves insulin resistance and β-cell function, leading to amelioration of glucose tolerance. Taken together, the JNK pathway plays a crucial role in the progression of insulin resistance as well as β-cell dysfunction and thus could be a potential therapeutic target for the "glucose toxicity" found in diabetes.
References
- Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 2001. 50:S154-S159. [DOD]
- Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes - a convergence of glucotoxicity and lipotoxicity. Endocrinology 2002. 143:339-342. [DOD] [CrossRef]
- Sharma A, Olson LK, Robertson RP, Stein R. The reduction of insulin gene transcription in HIT-T15beta-cells chronically exposed to high glucose concentration is associated with loss of RIPE3b1 and STF-1 transcription factor expression. Mol Endocrinol 1995. 9:1127-1134. [DOD] [CrossRef]
- Moran A, Zhang HJ, Olson LK, Harmon JS, Poitout V, Robertson RP. Differentiation of glucose toxicity from beta cell exhaustion during the evolution of defective insulin gene expression in the pancreatic islet cell line, HIT-T15. J Clin Invest 1997. 99:534-539. [DOD]
- Jonas JC, Sharma A, Hasenkamp W, Iikova H, Patane G, Laybutt R, Bonner-Weir S, Weir GC. Chronic hyperglycemia triggers loss of pancreatic beta-cell differentiation in an animal model of diabetes. J Biol Chem 1999. 274:14112-14121. [DOD] [CrossRef]
- Dandona P, Thusu K, Cook S, Snyder B, Makowski J, Armstrong D, Nicotera T. Oxidative damage to DNA in diabetes mellitus. Lancet 1996. 347:444-445. [DOD] [CrossRef]
- Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 1999. 48:1-9. [DOD]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000. 404:787-790. [DOD] [CrossRef]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001. 414:813-820. [DOD] [CrossRef]
- Kaneto H, Fujii J, Myint T, Islam KN, Miyazawa N, Suzuki K, Kawasaki Y, Nakamura M, Tatsumi H, Yamasaki Y, Taniguchi N. Reducing sugars trigger oxidative modification and apoptosis in pancreatic beta-cells by provoking oxidative stress through the glycation reaction. Biochem J 1996. 320:855-863. [DOD]
- Matsuoka T, Kajimoto Y, Watada H, Kaneto H, Kishimoto M, Umayahara Y, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest 1997. 99:144-150. [DOD]
- Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997. 46:1733-1742. [DOD]
- Ihara Y, Toyokuni S, Uchida K. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 1999. 48:927-932. [DOD]
- Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y, Hori M. Beneficial effects of antioxidants for diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999. 48:2398-2406. [DOD]
- Tanaka Y, Gleason CE, Tran POT, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci USA 1996. 96:10857-10862. [DOD] [CrossRef]
- Kaneto H, Kajimoto Y, Fujitani Y, Matsuoka T, Sakamoto K, Matsuhisa M, Yamasaki Y, Hori M. Oxidative stress induces p21 expression in pancreatic islet cells: possible implication in beta-cell dysfunction. Diabetologia 1999. 42:1093-1097. [DOD] [CrossRef]
- Kajimoto Y, Matsuoka T, Kaneto H, Watada H, Fujitani Y, Kishimoto M, Sakamoto K, Matsuhisa M, Kawamori R, Yamasaki Y, Hori M. Induction of glycation suppresses glucokinase gene expression in HIT-T15 cells. Diabetologia 1999. 42:1417-1424. [DOD] [CrossRef]
- Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem 1999. 274:27905-27913. [DOD] [CrossRef]
- Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function by provoking oxidative stress. J Biol Chem 2001. 276:31099-31104. [DOD] [CrossRef]
- Tanaka Y, Tran POT, Harmon J, Robertson RP. A role of glutathione peroxidase in protecting pancreatic beta-cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci USA 2002. 99:12363-12368. [DOD] [CrossRef]
- Gorogawa S, Kajimoto Y, Umayahara Y, Kaneto H, Watada H, Kuroda A, Kawamori D, Yasuda T, Matsuhisa M, Yamasaki Y, Hori M. Probucol preserves pancreatic beta-cell function through reduction of oxidative stress in type 2 diabetes. Diabetes Res Clin Prac 2002. 57:1-10. [DOD] [CrossRef]
- Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamura K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M, Araki E. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun 2003. 300:216-222. [DOD] [CrossRef]
- Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003. 52:581-587. [DOD]
- Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004. 53:S119-S124. [DOD]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signalling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003. 52:1-8. [DOD]
- Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing-transactivator of the insulin gene. EMBO J 1993. 12:4251-4259. [DOD]
- Leonard J, Peers B, Johnson T, Ferreri K, Lee S, Montminy MR. Characterization of somatostatin transactivating factor-1, a novel homeobox factor that stimulates somatostatin expression in pancreatic islet cells. Mol Endocrinol 1993. 7:1275-1283. [DOD] [CrossRef]
- Miller CP, McGehee RE, Habener JF. IDX-1: a new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J 1994. 13:1145-1156. [DOD]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994. 37:606-609. [DOD] [CrossRef]
- Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, Teitelman G. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development 1995. 121:11-18. [DOD]
- Slack JMW. Developmental biology of the pancreas. Development 1995. 121:1569-1580. [DOD]
- Offield MF, Jetton TL, Labosky P, Ray M, Stein R, Magnuson M, Hogan BLM, Wright CVE. PDX-1 is required for pancreas outgrowth and differentiation of the rostral duodenum. Development 1996. 122:983-985. [DOD]
- Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 1996. 122:1409-1416. [DOD]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 1997. 15:106-110. [DOD] [CrossRef]
- Kaneto H, Miyagawa J, Kajimoto Y, Yamamoto K, Watada H, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y, Higashiyama S, Taniguchi N. Expression of heparin-binding epidermal growth factor-like growth factor during pancreas development: a potential role of PDX-1 in transcriptional activation. J Biol Chem 1997. 272:29137-29143. [DOD] [CrossRef]
- Sander M, German MS. The beta cell transcription factors and development of the pancreas. J Mol Med 1997. 75:327-340. [DOD] [CrossRef]
- Stoffers DA, Heller RS, Miller CP, Habener JF. Developmental expression of the homeodomain protein IDX-1 mice transgenic for an IDX-1 promoter/LacZ transcriptional reporter. Endocrinology 1999. 140:5374-5381. [DOD]
- Holland AM, Hale MA, Kagami H, Hammer RE, MacDonald RJ. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci USA 2002. 99:12236-12241. [DOD] [CrossRef]
- Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets expanded ductal tissue. Proc Natl Acad Sci USA 2000. 97:7999-8004. [DOD] [CrossRef]
- Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I, Barshack I, Seijffers R, Kopolovic J, Kaiser N, Karasik A. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nature Med 2000. 6:568-572. [DOD] [CrossRef]
- Kojima H, Nakamura T, Fujita Y, Kishi A, Fujimiya M, Yamada S, Kudo M, Nishio Y, Maegawa H, Haneda M, Yasuda H, Kojima I, Seno M, Wong NCW, Kikkawa R, Kashiwagi A. Combined expression of pancreatic duodenal homeobox 1 and islet factor 1 induces immature enterocytes to produce insulin. Diabetes 2002. 51:1398-1408. [DOD]
- Yoshida S, Kajimoto Y, Yasuda T, Watada H, Fujitani Y, Kosaka H, Gotow T, Miyatsuka T, Umayahara Y, Yamasaki Y, Hori M. PDX-1 induces differentiation of intestinal epithelioid IEC-6 into insulin-producing cells. Diabetes 2002. 51:2505-2513. [DOD]
- Ber I, Shternhall K, Perl S, Ohanuna Z, Goldberg I, Barshack I, Benvenisti-Zarum L, Meivar-Levy I, Ferber S. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem 2003. 22:31950-31957. [DOD] [CrossRef]
- Miyatsuka T, Kaneto H, Kajimoto Y, Hirota S, Arakawa Y, Fujitani Y, Umayahara Y, Watada H, Yamasaki Y, Magnuson MA, Miyazaki J, Hori M. Ectopically expressed PDX-1 in liver initiates endocrine and exocrine pancreas differentiation but causes dysmorphogenesis. Biochem Biophys Res Commun 2003. 310:1017-1025. [DOD] [CrossRef]
- Noguchi H, Kaneto H, Weir GC, Bonner-Weir S. PDX-1 protein containing its own Antennapedia-like protein transduction domain can transduce pancreatic duct and islet cells. Diabetes 2003. 52:1732-1737. [DOD]
- Moritoh Y, Yamato E, Yasui Y, Miyazaki S, Miyazaki J. Analysis of insulin-producing cells during in vitro differentiation from feeder-free embryonic stem cells. Diabetes 2003. 52:1163-1168. [DOD]
- Taniguchi H, Yamato E, Tashiro F, Ikegami H, Ogihara T, Miyazaki J. Beta-cell neogenesis induced by adenovirus-mediated gene delivery of transcription factor pdx-1 into mouse pancreas. Gene Ther 2003. 10:15-23. [DOD] [CrossRef]
- Tang DQ, Cao LZ, Burkhardt BR, Xia CQ, Litherland SA, Atkinson MA, Yang L-J. In vivo and in vitro characterization of insulin-producing cells obtained from murine bone marrow. Diabetes 2004. 53:1721-1732. [DOD]
- Leon-Quinto T, Jones J, Skoudy A, Burcin M, Soria B. In vitro directed differentiation of mouse embryonic stem cells into insulin-producing cells. Diabetologia 2004. 47:1442-1451. [DOD]
- Miyazaki S, Yamato E, Miyazaki J. Regulated expression of pdx-1 promotes in vitro differentiation of insulin-producing cells from embryonic stem cells. Diabetes 2004. 53:1030-1037. [DOD]
- Koizumi M, Doi R, Toyoda E, Tulachan SS, Kami K, Mori T, Ito D, Kawaguchi Y, Fujimoto K, Gittes GK, Imamura M. Hepatic regeneration and enforced PDX-1 expression accelerate transdifferentiation in liver. Surgery 2004. 136:449-457. [DOD] [CrossRef]
- Petersen HV, Serup P, Leonard J, Michelsen BK, Madsen OD. Transcriptional regulation of the human insulin gene is dependent on the homeodomain protein STF1/IPF1 acting through the CT boxes. Proc Natl Acad Sci USA 1994. 91:10465-10469. [DOD]
- Peers B, Leonard J, Sharma S, Teitelman G, Montminy MR. Insulin expression in pancreatic islet cells relies on cooperative interactions between the helix loop helix factor E47 and the homeobox factor STF-1. Mol Endocrinol 1994. 8:1798-1806. [DOD] [CrossRef]
- Waeber G, Thompson N, Nicod P, Bonny C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol Endocrinol 1996. 10:1327-1334. [DOD] [CrossRef]
- Watada H, Kajimoto Y, Umayahara Y, Matsuoka T, Kaneto H, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y. The human glucokinase gene beta-cell-type promoter: An essential role of insulin promoter factor 1 (IPF1)/PDX-1 in its activation in HIT-T15 cells. Diabetes 1996. 45:1478-1488. [DOD]
- Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev 1998. 12:1763-1768. [DOD]
- Wang H, Maechler P, Ritz-Laser B, Hagenfeldt KA, Ishihara H, Philippe J, Wollheim CB. Pdx1 level defines pancreatic gene expression pattern and cell lineage differentiation. J Biol Chem 2001. 276:25279-25286. [DOD] [CrossRef]
- Leibowitz G, Ferber S, Apelqvist A, Edlund H, Gross DJ, Cerasi E, Melloul D, Kaiser N. IPF1/PDX1 deficiency and beta-cell dysfunction in psammomys obesus, an animal with type 2 diabetes. Diabetes 2001. 50:1799-1806. [DOD]
- McKinnon CM, Docherty K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001. 44:1203-1214. [DOD] [CrossRef]
- Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, Wright CV, Powers AC. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem 2002. 277:11225-11232. [DOD]
- Chakrabarti SK, James JC, Mirmira RG. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, pdx1: importance of chromatin structure in directing promoter binding. J Biol Chem 2002. 277:13286-13293. [DOD] [CrossRef]
- Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest 2004. 114:828-836. [DOD] [CrossRef]
- Dutta S, Bonner-Weir S, Montminy M, and Wright C. Regulatory factor linked to late-onset diabetes? Nature 1998. 392:560. [DOD]
- Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 1997. 17:138-139. [DOD] [CrossRef]
- Hibi M, Lin A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 1993. 7:2135-2148. [DOD]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kianse stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994. 76:1025-1037. [DOD] [CrossRef]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000. 103:239-252. [DOD] [CrossRef]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001. 410:37-40. [DOD] [CrossRef]
- Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 2002. 277:30010-30018. [DOD] [CrossRef]
- Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, Watada H, Leibiger IB, Yamasaki Y, Hori M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun N-terminal kinase. Diabetes 2003. 52:2896-2904. [DOD]
- Corbett JA, Wang JL, Sweetland MA, Lancaster JR Jr, McDaniel ML. Interleukin 1 beta induces the formation of nitric oxide by beta-cells purified from rodent islets of Langerhans: evidence for the beta-cell as a source and site of action of nitric oxide. J Clin Invest 1992. 90:2384-2391. [DOD]
- Eizirik DL, Sandler S, Welsh N, Cetkovic-Cvrlje M, Nieman A, Geller DA, Pipeleers DG, Bendtzen K, Hellerstrom C. Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J Clin Invest 1994. 93:1968-1974. [DOD]
- Ankarcrona M, Dypbukt JM, Brune B, Nicotera P. Interleukin-1beta-induced nitric oxide production activates apoptosis in pancreatic RINm5F cells. Exp Cell Res 1994. 213:172-177. [DOD] [CrossRef]
- Kaneto H, Fujii J, Seo HG, Suzuki K, Matsuoka T, Nakamura N, Tatsumi H, Yamasaki Y, Kamada T, Taniguchi N. Apoptotic cell death triggered by nitric oxide in pancreatic beta-cells. Diabetes 1995. 44:733-738. [DOD]
- Ammendrup A, Maillard A, Nielsen K, Aabenhus Andersen N, Serup P, Dragsbaek Madsen O, Mandrup-Poulsen T, Bonny C. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 2000. 49:1468-1476. [DOD]
- Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Waeber G. IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J Biol Chem 2000. 275:16466-16472. [DOD] [CrossRef]
- Mandrup-Poulsen T. Beta-cell apoptosis: stimuli and signaling. Diabetes 2001. 50:S58-S63. [DOD]
- Bonny C, Oberson A, Negri S, Sause C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 2001. 50:77-82. [DOD]
- Bonny C, Nicod P, Waeber G. IB1, a JIP-1-related nuclear protein present in insulin-secreting cells. J Biol Chem 1998. 273:1843-1846. [DOD] [CrossRef]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 1997. 277:693-696. [DOD]
- Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, Hani EH, Vionnet N, Nicod P, Boutin P, Froguel P. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet 2000. 24:291-295. [DOD] [CrossRef]
- Nakatani Y, Kaneto H, Kawamori D, Hatazaki M, Miyatsuka T, Matsuoka T, Kajimoto Y, Matsuhisa M, Yamasaki Y, Hori M. Modulation of the JNK pathway in liver affects insulin resistance status. J Biol Chem 2004. 279:45803-45809. [DOD] [CrossRef]
- Hirosumi J, Tuncman G, Chang L, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature 2002. 420:333-336. [DOD] [CrossRef]
- Aguirre V, Davis R, White MF. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J Biol Chem 2000. 275:9047-9054. [DOD] [CrossRef]
- Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Dis 2003. 2:554-565. [DOD]
- Bennett BL, Satoh Y, Lewis A. JNK: a new therapeutic target for diabetes. Curr Opin Pharm 2003. 3:420-425. [DOD] [CrossRef]
- Schwarze SR, Ho A, Vocero-Akbani AM, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999. 285:1569-1572. [DOD] [CrossRef]
- Elliott G, O'Hare P. Intracellular trafficking and protein delivery by a herpesvirus structure protein. Cell 1997. 88:223-233. [DOD] [CrossRef]
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988. 55:1189-1193. [DOD] [CrossRef]
- Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p-27Kip1 induces cell migration. Nature Med 1998. 4:1449-1452. [DOD] [CrossRef]
- Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, Wender PA, Khavari PA. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nature Med 2000. 6:1253-1257. [DOD] [CrossRef]
- Noguchi H, Kaneto H, Weir GC, Bonner-Weir S. PDX-1 protein containing its own Antennapedia-like protein transduction domain can transduce pancreatic duct and islet cells. Diabetes 2003. 52:1732-1737. [DOD]
- Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, Li ST, Kobayashi N, Matsumoto S, Tanaka K, Tanaka N, Matsui H. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nature Med 2004. 10:305-309. [DOD] [CrossRef]
- Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka T, Matsuhisa M, Kajimoto Y, Ichijo H, Yamasaki Y, Hori M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nature Med 2004. 10(10):1128-1132. [DOD]
This article has been cited by other articles:

|
Obesity, metabolic syndrome, adipocytes and vascular function: A holistic viewpoint
Achike FI, To NH, Wang H, Kwan CY
Clin Exp Pharmacol Physiol 2011. 38(1):1-10
|
|

|
Role of the transcriptional factor C/EBPβ in free fatty acid-elicited β-cell failure
Plaisance V, Perret V, Favre D, Abderrahmani A, Yang JY, Widmann C, Regazzi R
Mol Cell Endocrinol 2009. 305(1-2):47-55
|
|

|
The effects of taurine, taurine homologs and hypotaurine on cell and membrane antioxidative system alterations caused by type 2 diabetes in rat erythrocytes
Gossai D, Lau-Cam CA
Adv Exp Med Biol 2009. 643:359-368
|
|

|
Chromium (VI) induces insulin resistance in 3T3-L1 adipocytes through elevated reactive oxygen species generation
Ge X, Liu Z, Qi W, Shi X, Zhai Q
Free Radic Res 2008. 42(6):554-563
|
|

|
Peripheral blood gene expression profiling for cardiovascular disease assessment
Aziz H, Zaas A, Ginsburg GS
Genomic Med 2007. 1(3-4):105-112
|
|

|
Type 2 Diabetes - Failure, Blame and Guilt in the Adoption of Insulin Therapy
Phillips P
Rev Diabet Stud 2005. 2(1):35-39
|
|

|
Tolerance Induction and Endogenous Regeneration of Pancreatic Beta-Cells in Established Autoimmune Diabetes
Sia C, Homo-Delarche F
Rev Diabet Stud 2004. 1(4):198-206
|
|
|


)
)
)
)

