Review
| Rev Diabet Stud,
2013,
10(2-3):191-203 |
DOI 10.1900/RDS.2013.10.191 |
Atherogenic Dyslipidemia and Combination Pharmacotherapy in Diabetes: Recent Clinical Trials
Sandra J. Hamilton1, Gerald F. Watts2
1Combined Universities Centre for Rural Health, University of Western Australia, Geraldton, Australia
2School of Medicine and Pharmacology, Royal Perth Hospital Unit, University of Western Australia, Perth, Australia
Address correspondence to: Gerald F. Watts, GPO Box X2213, Perth, Western Australia, Australia 6847, e-mail: gerald.watts@uwa.edu.au
Manuscript submitted March 20, 2013; resubmitted June 19, 2013; accepted June 19, 2013.
Keywords: atherogenic dyslipidemia, clinical trials, pharmacotherapy, type 2 diabetes, fatty acid, lipoprotein, HDL cholesterol
Abstract
Patients with type 2 diabetes (T2D) are at a markedly increased risk of cardiovascular disease (CVD). Dyslipidemia is a common risk factor and a strong predictor of CVD in T2D patients. Although statins decrease the incidence of CVD in T2D, residual cardiovascular risk remains high despite the achievement of optimal or near-optimal plasma low-density lipoprotein (LDL) cholesterol concentrations. This may, in part, be due to uncorrected atherogenic dyslipidemia. Hypertriglyceridemia, the driving force behind diabetic dyslipidemia, results from hepatic overproduction and/or delayed clearance of triglyceride-rich lipoproteins. In patients treated with a statin to LDL-cholesterol goals, the addition of ezetimibe, fenofibrate, niacin, or n-3 fatty acid ethyl esters may be required to correct the persistent atherogenic dyslipidemia. Clinical trial evidence describing best practice is limited, but recent data supports the strategy of adding fenofibrate to a statin, and suggests specific benefits in dyslipidemic patients and in the improvement of diabetic retinopathy. However, based on results from a recent clinical trial, niacin should not be added to a statin in individuals with low high-density lipoprotein cholesterol and very well controlled LDL-cholesterol. Further evidence is required to support the role of ezetimibe and n-3 fatty acids in treating residual CVD risk in statin-treated T2D patients.
Abbreviations: ACCORD - Action to Control Cardiovascular Risk in Diabetes; AHEAD - Action for Health in Diabetes; AIM-HIGH - Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes; Apo - apolipoprotein; ARBITER - Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol; CAD - coronary artery disease; CDP - Coronary Drug Project; CETP - cholesterylester transfer protein; CIMT - carotid intima-media thickness; CTT - Collaborative Treatment Trialists; CVD - cardiovascular disease; DGAT-2 - diacylglycerol acyl transferase; DHA - docosahexemoic acid; ED - endothelial dysfunction; EPA - eicosapentenoic acid; ER - extended release; FFA - free fatty acid; FIELD - Fenofibrate Intervention and Event Lowering in Diabetes; GISSI-Prevenzione - Gruppo Italian per lo Studio della Sopravvivenza nell'Infarto Miocardio GISSI-Prevenzione; HALTS - ARBITER-6-HDL and LDL Treatment Strategies in Atherosclerosis; HATS - HDL-Atherosclerosis Treatment Study; HDL - high-density lipoprotein; HPS2-THRIVE - Heart Protection Study-2 and the Treatment of HDL to reduce the incidence of vascular events; IHD - ischemic heart disease; JELIS - Japan Eicosapentenoic Acid Lipid Intervention Study; LDL - low-density lipoprotein; LpA Lipoprotein A; LPL - lipoprotein lipase; NCEP-ATP III - National Cholesterol Education Program Adult Treatment Panel III; ORIGIN - Outcome Reduction with an Initial Glargine Intervention; PLPT - phospholipid transfer protein; PPAR-α - Peroxisome proliferator-activated receptors α; PROVE IT-TIMI - Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction; REDUCE-IT - A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients with Hypertriglyceridemia and on Statin; SANDS - Stop Atherosclerosis in Native Diabetes Study; sdLDL - Small dense low-density lipoprotein; T2D - type 2 diabetes; TLC - therapeutic lifestyle change; TRL - triglyceride-rich lipoprotein; VLDL - very low-density lipoprotein
1. Introduction
Dyslipidemia is a common risk factor and powerful predictor of cardiovascular disease (CVD) in high-risk patients, including those with type 2 diabetes (T2D) [1, 2]. Although low-density lipoprotein (LDL) cholesterol is the main target for treatment [3, 4], clinical trials consistently demonstrate high residual cardiovascular risk in statin-treated patients [2]. This may be because statins incompletely correct the vascular risk that is attributable to atherogenic dyslipidemia [2]. With a focus on T2D patients, we review in this article recent clinical trials employing therapeutic agents for the treatment of atherogenic dyslipidemia.
2. Atherogenic dyslipidemia
Diabetic dyslipidemia involves a cluster of lipid and lipoprotein abnormalities [5]. Elevated plasma concentrations of triglycerides and reduced high-density lipoprotein cholesterol (HDL-cholesterol), in both the fasting and postprandial states, are the core lipoprotein abnormalities [5]. The accumulation of small dense low-density lipoprotein (sdLDL) particles and triglyceride-rich lipoproteins (TRLs), including chylomicron remnants and very-low-density lipoprotein (VLDL) remnants, are also characteristic of the atherogenic lipid profile [5-8]. These abnormalities are reflected by increased plasma concentrations of non-HDL cholesterol and apolipoprotein B-100 (apoB) [6].
In the postprandial state, there is an increase in plasma TRLs and their remnants and qualitative changes in LDL and HDL particles [5]. Therefore, hypertriglyceridemia is a marker of a range of lipoprotein abnormalities not routinely measured in clinical practice [6]. Evidence suggests that fasting and non-fasting plasma triglyceride concentrations, and by implication triglyceride-rich apoB-containing remnant lipoproteins, are strong predictors of CVD [6-8]. Utilizing Mendelian randomization, two recent studies have demonstrated a causal association between genetically increased remnant cholesterol in hypertriglyceridemia, particularly due to genetic variation in the apoA5 and LPL genes, and an increased risk of ischemic heart disease (IHD) [9, 10]. Indeed, a 1 mmol/l increase in non-fasting remnant cholesterol was associated with a 2.8-fold increase in causal risk for ischemic heart disease (IHD), independently of a reduced HDL-cholesterol [10]. The association with apoA5 gene variants is supported by a large meta-analysis [8]. However, the evidence for a causal association between low HDL-cholesterol and atherogenesis is less compelling. Findings from a recent Mendelian randomization study challenge the concept that HDL-cholesterol raising translates to reduced risk of coronary artery disease (CAD) [11]. In the dal-OUTCOMES study, dalcetrapib, a cholesteryl ester transfer protein (CETP) inhibitor, raised HDL-cholesterol levels, but did not reduce recurrent cardiovascular events in patients with recent acute coronary syndrome [12].
3. Pathogenesis of atherogenic dyslipidemia in type 2 diabetes
Atherogenic dyslipidemia is seen in most patients with elevated triglycerides >2.2 mmol/l and reduced HDL-cholesterol <1.0 mmol/l. The majority of these patients have T2D, central adiposity, or insulin resistance [5, 6].
Etiology. The etiology of diabetic dyslipidemia is complex, and hypertriglyceridemia is central to its pathogenesis [6, 13, 14]. Diabetic dyslipidemia relates collectively to hyperglycemia [15], insulin resistance [15], hyperinsulinemia [15, 16], abdominal visceral adipose disposition, increased liver fat content [16], and dysregulated fatty acid metabolism [16]. Insulin resistance increases fatty acid flux from visceral adipose tissue to the liver, inducing hepatic steatosis, oversecretion of larger triglyceride-rich VLDL1 particles into the plasma [5, 6, 16], and a reduction in the inhibitory effect of insulin on hepatic apoB secretion [17-19]. Hyperglycemia also drives the overproduction of VLDL1, in particular increased VLDL1 triglyceride production rate [15]. Collectively, plasma glucose, insulin, and free fatty acids explain approximately half of the variation in VLDL1 production rate [15].
Impaired chylomicron clearance in T2D results from the reduced activity of lipoprotein lipase (LPL), an endothelial bound enzyme, and decreased receptor-mediated endocytosis in the liver [5, 6, 20, 21]. VLDL1 particles compete with chylomicrons and its remnants for clearance by saturating the lipolytic capacity of LPL and the activity of hepatic receptors, thereby increasing postprandial dyslipidemia [5, 6, 20]. Hepatic secretion of apolipoprotein CIII (apoCIII) is also increased in insulin resistance. This small protein, which is attached to VLDL, contributes to the delayed clearance of TRLs by inhibiting LPL and the binding of remnant TRLs to hepatic clearance receptors [6, 20]. These mechanisms collectively account for postprandial lipemia [5, 20], and may be an important causal mechanism of endothelial dysfunction (ED) in T2D; they may be treatment targets for reducing residual cardiovascular risk.
3.1 Atherogenicity
Important compositional and atherogenic changes in lipoproteins are seen in T2D [5]. An increased VLDL triglyceride pool leads to cholesterol depletion and triglyceride enrichment of LDL and HDL, mediated via the action of cholesterol ester transfer protein (CETP) [5, 6]. Increased phospholipid transfer protein (PLTP) activity may contribute to hypertriglyceridemia and compositional changes in HDL. Furthermore, the overactivity of hepatic lipase, which is commonly elevated in T2D, increases the lipolysis of triglyceride-enriched LDL and HDL particles [22, 23]. Compositional changes in HDL are also mediated by the actions of LPL [5]. Collectively, these compositional changes produce smaller and denser lipoprotein particles that are potentially more atherogenic. Small dense LDL particles more easily penetrate the arterial wall, and have a higher binding affinity to intimal proteoglycans than more buoyant larger LDL particles [5, 22, 24, 25]. In the intima, retained LDL particles are modified when exposed to oxidative stress, with sdLDL having an increased sensitivity to oxidation; glycation of LDL further increases this susceptibility to oxidation [5].
Diabetic dyslipidemia is also characterized by low HDL-cholesterol concentrations with greater reductions in HDL2 than HDL3 [5]. In parallel with these reductions in HDL particles are reductions in plasma levels of apolipoprotein A-I (apoA-I) and apoA-II and HDL lipoproteins containing both Apo A-I and Apo A-II (LpA-I:A-II) [5, 22, 26]. These compositional changes in HDL particles are important with respect to endothelial dysfunction (ED) and atherogenicity, as they are associated with the reduction in rates of reverse cholesterol transport and a decrease in the direct anti-atherogenic effects of HDL, including its antioxidant, anti-inflammatory, and anti-thrombotic effects [22, 27-29].
3.2 Targeting atherogenic dyslipidemia
Treatment for atherogenic dyslipidemia should focus on the progressing compositional changes in lipoprotein particles, aiming to reduce hepatic secretion of VLDL-apoB and -triglyceride and transfer of apoB from VLDL to LDL [6]. It should also aim to accelerate the clearance of all apoB-containing lipoproteins [6, 20]. Lifestyle changes are essential and should include low fat diets, weight loss, increased physical activity, and reduced sedentary time [6, 20]. However, pharmacotherapy is often required with statins as first line therapy [20, 30], but statins may not adequately correct the metabolic abnormalities. Therefore, combination therapies may be required [30].
4. Guideline for the management of atherogenic dyslipidemia
Several guidelines provide evidence-based recommendations for addressing diabetic dyslipidemia [3, 4, 31-35]. Two recent reports focus more specifically on elevated triglycerides and low HDL-cholesterol [36, 37]. Table 1 summarizes the recommended treatment targets for diabetic dyslipidemia. In T2D, LDL-cholesterol lowering remains the primary focus of therapeutic interventions [3, 4, 31, 32]. T2D patients with overt CVD or high cardiovascular risk should have statin therapy and therapeutic lifestyle changes (TLCs) initiated regardless of baseline lipid levels. In lower risk patients, statin therapy should be initiated if LDL-cholesterol levels remain above 2.6 mmol/l following TLC efforts or in those with multiple CVD risk factors [31]. These recommendations are supported by evidence of CVD reduction in diabetic patients in large outcome-based clinical trials [38-41].
Table
1.
Recommended treatment targets for diabetic dyslipidemia |
|
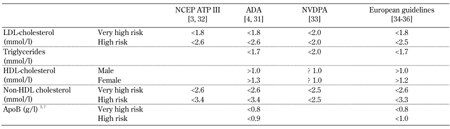 |
 |
Legend:
NCEP ATP III - Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III); ADA - American Diabetes Association; NVDPA - National Vascular Disease Prevention Alliance of Australia. |
|
If LDL-cholesterol reduction is inadequate with a maximum tolerated statin dose, then adding a second therapeutic agent (ezetimibe, fibrate, or niacin) may be required [31]. For patients with elevated triglycerides (>2.3 mmol/l), the use of non-HDL cholesterol as a secondary treatment target is recommended [3, 32, 36, 37]. ApoB, a measure of LDL particle number is also a recommended treatment target in patients at cardiometabolic risk [4, 35, 37]. Recommendations for these patients include combination therapy with a second lipid-regulating agent (fibrate, niacin, or omega-3 fatty acids) or intensification of LDL-cholesterol lowering [3, 4, 36]. Evidence from the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid study supports the use of combined statin and fenofibrate therapy in hypertriglyceridemic T2D patients [42].
5. Therapeutic regulation of atherogenic dyslipidemia in type 2 diabetes
5.1 Monotherapies
Lifestyle interventions. Initial management should include a personalized lifestyle modification program to optimize weight loss, lipids, and glycemic control. Evidence suggests that lifestyle programs aimed at achieving weight loss improve many of the metabolic abnormalities in T2D that are associated with ED and CVD, such as hyperglycemia, insulin resistance, visceral obesity, hypertension, and dyslipidemia. Weight loss improves the lipoprotein dysregulation in obese subjects with metabolic syndrome and T2D [20, 43-45]. Benefits of weight reduction in T2D increase steadily with increasing weight loss and include reductions in waist circumference, blood pressure, fasting glucose, HbA1c, and serum triglycerides, resulting in improved metabolic control and CVD risk factor reduction [46, 47].
In the Look AHEAD (Action for Health in Diabetes) study, weight loss and improved physical fitness in type 2 diabetic patients was associated with improved glycemic control and CVD risk factor reduction [47, 48]. After 4 years of follow-up in the Look AHEAD study, intensive lifestyle intervention was associated with greater weight loss and improvements in physical fitness [48]. Improvements in glycemic control, blood pressure, triglycerides, and HDL-cholesterol were also demonstrated [48]. In T2D patients, a reduction in insulin resistance and fat mass following prolonged aerobic exercise resulted in an improved lipoprotein metabolism [49].
Should dyslipidemia persist following a trial of intensified lifestyle changes, the next approach is pharmacotherapy, either an intensification of statin therapy or the addition of a second lipid regulating agent.
Lipid regulating therapy. (i) Statin monotherapy: Patients with an increasing number of metabolic syndrome components, with or without diabetes, have a progressive risk of CVD, and derive greater incremental benefit from higher dose statin therapy [50]. Statins, the most potent agents for lowering plasma LDL-cholesterol and apoB concentrations, have a less potent but significant effect on reducing plasma triglycerides [6]. Indeed, statin therapy in hypertriglyceridemic patients, with and without T2D, has been shown to reduce triglyceride concentrations by up to 45% [51-55], in a dose-dependent manner and proportional to LDL-cholesterol lowering [54, 55]. Statin-treated patients with combined low LDL-cholesterol (<1.8 mmol/l) and low triglyceride (<1.7 mmol/l) levels had the lowest coronary heart disease (CHD) event rate in the PROVE IT-TIMI 22 trial [56]. Evidence suggests that statins may mediate triglyceride-lowering in T2D by increasing the catabolism of TRL- triglyceride [52] and TRL's VLDL1-ApoB, VLDL2-ApoB, and IDL-ApoB [57]. The more potent statin, rosuvastatin, has been shown to further mediate triglyceride lowering by reducing the production rate and secretion of VLDL1-ApoB [57]. However, this effect has not consistently been shown with statin therapy, indicating that limiting de novo cholesterol synthesis is insufficient for reducing the effects of insulin resistance on hepatic VLDL production [6, 20, 58, 59]. Hence, targeting the hepatic production of triglycerides by additional measures such as lifestyle or other pharmacotherapy is required to optimize the management of dyslipidemia in T2D.
However, several large clinical trials have shown that statins decrease cardiovascular events in T2D patients, attaining an average 20% reduction in CVD events per 1 mmol/l decrease in LDL-cholesterol [41]. In coronary patients with T2D or with the metabolic syndrome alone, high-dose statin (atorvastatin 80 mg daily) decreased the relative risk of CVD by 25-30% compared with lower-dose statin (atorvastatin 10 mg daily) [60]. Evidence also supports the use of statins in T2D patients without CVD, but with at least one other cardiovascular risk factor, including hypertension [39, 61]. These findings are consistent with the Heart Protection Study (HPS) [38] and the Collaborative Treatment Trialists' (CTT) Collaborators meta-analysis of statin trials in 18,686 people with diabetes [41]. The cardiovascular benefits of statins relate primarily to the lowering of LDL-cholesterol and lipoprotein remnants, but residual cardiovascular risk in statin-treated patients remains high [2, 41]. In T2D, residual cardiovascular risk extends to both macroangiopathy and microangiopathy [2].
(ii) Fibric acid derivatives: A meta-analysis has concluded that fibrates are effective in reducing CVD events, primarily by prevention of coronary events [62]. Another recent meta-analysis also suggested that fibrates may be particularly useful in improving dyslipidemia and preventing CVD in people with mild to moderate chronic kidney disease, including diabetics [63]. The lipid-regulating effects of fibrates, mediated via the peroxisome proliferator-activated alpha (PPAR-α) receptor, predominantly promote fatty acid catabolism and reverse cholesterol transport, resulting in triglyceride lowering and increased HDL-cholesterol and LDL particle size [64]. Fibrates can lower plasma triglyceride and LDL-cholesterol concentrations by up to 50% and 20%, respectively. Fibrates also increase HDL-cholesterol by up to 20%, and enhance the formation of large, less dense LDL-particles [42]. Clinical trials also confirm the benefits of fibrates in T2D patients [42, 65-67]. In a subgroup analysis of the Helsinki Heart Study, diabetic patients, when compared with non-diabetic subjects, were more dyslipidemic, at higher CVD risk, and achieved a modest but non-significant reduction in CVD risk with gemfibrozil therapy [65]. Although the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study did not select T2D patients for having atherogenic dyslipidemia, a post-hoc analysis of patients with triglycerides ≥2.3 mmol/l demonstrated that fenofibrate reduced CVD events in hypertriglyceridemic T2D patients with or without low HDL-cholesterol [67]. The greatest benefit was seen in those patients with triglycerides ≥2.3 mmol/l and reduced HDL-cholesterol (<1.0 mmol/l); a relative risk reduction of 27% was demonstrated [67]. In FIELD, improvement in microangiopathy was also demonstrated, reflected by reductions in albuminuria [68], laser photocoagulation [69], and minor amputations [70]. However, the aforementioned evidence is confined to T2D patients not taking background statin therapy. Evidence for the effects of combination statin plus fibrate therapy on CVD outcomes is currently limited to the ACCORD study. ACCORD recently reported on the efficacious effect of adding fenofibrate to ongoing simvastatin therapy on CVD events in T2D patients [42]. ACCORD will be discussed in a subsequent section.
(iii) Nicotinic acid (niacin): At therapeutic doses, niacin exerts a global improvement in lipid and lipoprotein metabolism, and remains the most efficacious therapy available for increasing HDL-cholesterol [30, 71, 72]. Niacin has been shown to decrease plasma triglycerides and LDL-cholesterol by up to 35% and 15%, respectively, and increase HDL-cholesterol by up to 30% in a dose dependent manner [30]. Many of niacin's effects are thought to derive from its action on adipose tissue [73]. However, the cellular mechanism for niacin's lipid-lowering effects were not fully elucidated until the identification of a G protein-coupled receptor GPR109A (HM74A) in 2003, which is highly expressed in adipose tissue, acts as a high-affinity receptor for nicotinic acid, and mediates antilipolytic effects [72, 74-77]. By binding to GPR109A, niacin inhibits hormone-sensitive lipase activity, resulting in decreased free fatty acid (FFA) release from adipose tissues. This results in a decreased flux of FFA to the liver that may reduce triglyceride production and subsequent hepatic VLDL production [45, 74, 78]. Niacin may also directly and noncompetitively inhibit hepatic diacylglycerol acyl transferase (DGAT-2), the key enzyme in triglyceride synthesis [45, 79]. A mechanism for niacin's HDL raising effect remains to be fully elucidated, but it may involve the downregulation of the HDL-catabolism receptor, resulting in delayed catabolism of larger HDL particles [71, 78, 80, 81]. Furthermore, kinetic studies suggest that niacin may increase the secretion of HDL-apoA-1 [82]. The lipid-regulating effects of niacin may be further mediated through PPAR-mediated transcriptional regulation, and may involve all three PPAR isoforms, alpha (α), gamma (γ) and delta (δ) [73].
It was suggested in the Coronary Drug Project (CDP) that niacin monotherapy may decrease cardiovascular events and mortality [6, 30, 83, 84], with post-trial follow-up demonstrating that the benefits were independent of hyperglycemia, metabolic syndrome, and diabetes [6, 30, 85, 86]. This is important as it has been suggested that niacin impairs glucose/insulin homeostasis [87]. Niacin significantly improves diabetic dyslipidemia [88], and any deleterious effects on glycemic control can be offset by adjusting antidiabetic therapy [89]. The effects of niacin on background statin therapy is discussed in a subsequent section.
(iv) N-3 fatty acid ethyl esters: Supplementation with n-3 fatty acid ethyl esters (eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)), dose dependently lowers plasma triglycerides, particularly in patients with hypertrigylceridemia [6, 20]. Doses of 3-4 g daily of EPA and DHA are required. Therefore, commercially available concentrates of n-3 fatty acid ethyl esters, such as omacor, are required for lowering plasma triglycerides [6]. These are FDA approved, as an adjunct to diet, to mitigate the risk of pancreatitis in patients with plasma triglycerides >5.5 mmol/l [6]. Improved triglyceride metabolism by n-3 fatty acids involves several mechanisms, including the transcriptional regulation of hepatic genes involved in lipogenesis and fatty acid oxidation [6, 90]. N-3 fatty acids decrease the hepatic secretion of large-size VLDL in patients with the metabolic syndrome and T2D, explaining the fall in triglycerides in those receiving a statin [6, 20, 91]. There is an increased conversion of VLDL to LDL, but the elevation of LDL-cholesterol can be counteracted by a statin [6, 20].
In the Gruppo Italian per lo Studio della Sopravvivenza nell'Infarto Miocardio GISSI-Prevenzione (GISSI-Prevenzione) study, combination EPA/DHA dosed at 1 g daily reduced all-cause mortality and sudden death in subjects with previous myocardial infarction [92]. N-3 fatty acid supplementation in the ORIGIN (Outcome Reduction with an Initial Glargine Intervention) trial is discussed in a subsequent section [93]. A comprehensive review of the cardiovascular effects of omega-3 fatty acids has recently been published [94].
5.2 Addressing residual cardiovascular risk with combination pharmacotherapy: recent clinical trials
High residual cardiovascular risk is evident in statin-treated T2D patients [39, 41]. This residual risk may relate to persistent abnormalities in TRLs and HDLs which are not fully corrected by statins [56, 95]. Increasing statins may partially improve these lipoprotein abnormalities, but increases the risk of musculoskeletal side effects [96], to which diabetic patients are prone [6]. Evidence for the addition of ezetimibe, fibrates, niacin, and n-3 fatty acid ethyl esters as adjunctive treatments for residual dyslipidemia and CVD risk are discussed below.
Statins and ezetimibe. Ezetimibe, an intestinal cholesterol absorption inhibitor, lowers LDL-cholesterol by 10 to 20%. However, its effects on triglycerides and HDL-cholesterol are minor [6, 97]. It does this chiefly by increasing the catabolism of LDL and possibly IDL-apoB-100 by upregulation of hepatic receptors. This relates to the impact of inhibiting intestinal cholesterol absorption on hepatic cholesterol content [6, 20]. Combination statin/ezetimibe therapy has complementary effects on the catabolism of apoB-containing lipoproteins through "dual inhibition" of cholesterol synthesis and absorption [6, 20]. In T2D patients, combination ezetimibe with lower-dose statin therapy achieved LDL-cholesterol and non-HDL-cholesterol targets more effectively than statin alone [98]. In the Stop Atherosclerosis in Native Diabetes Study (SANDS), lowering of LDL-cholesterol to aggressive targets with ezetimibe plus statin, or statin alone, achieved regression of carotid intima-media thickness (CIMT), a surrogate marker of atherosclerosis, in T2D patients [99]. This related to the reduction in LDL-cholesterol and not the minimal changes in triglycerides or HDL-cholesterol [99]. Hence, further evidence is required to support the role of ezetimibe in treating residual CVD risk in statin-treated T2D patients.
Statins and fibrates. A recent pooled analysis of two controlled studies reported on the benefits of rosuvastatin and fenofibrate, alone or in combination, in patients with mixed dyslipidemia [100]. Patients were stratified according to high, moderate, and low CVD risk based on National Cholesterol Education Program Adult Treatment Panel (NCEP ATP) III guidelines. Rosuvastatin alone or in combination had similar effects on patients achieving LDL-cholesterol goals. However, combination therapy had a more favorable effect on total atherogenic burden compared with fenofibrate alone; a greater percentage of moderate- and high-risk patients achieve non-HDL target levels and ApoB <0.9 g/l in the high risk group [100].
A limited number of controlled studies have reported on the effects of combination statin plus fibrate therapy on CVD outcomes. The benefits of combined fibrate and statin therapy was examined in the ACCORD study.
(i) Action to Control Cardiovascular Risk in Diabetes (ACCORD)-Lipid study: ACCORD-Lipid reported on the effects of adding fenofibrate to ongoing statin treatment in 5518 T2D patients who had documented CVD or a least two additional cardiovascular risk factors, on a background of good glycemic and blood pressure control [42]. Patients already at LDL-cholesterol target (~2.6 mmol/l on a mean dose of 22 mg/day of simvastatin) were randomized to fenofibrate (54-145 mg/day) plus simvastatin (20-40 mg/day) or simvastatin (20-40 mg/day) alone. However, as the ACCORD trial also targeted glycemic control, the lipid arm was not restricted to patients with atherogenic dyslipidemia. Compared to simvastatin alone, 4.7 years of combination therapy did not reduce the rate of major fatal or nonfatal cardiovascular events. Compared with simvastatin alone, fenofibrate significantly reduced plasma triglycerides (-22% vs. -8.7%) and, although modest, a significant increase in HDL-cholesterol (+8.4% vs. +6.0%) was demonstrated. No significant differences were reported in LDL-cholesterol, which averaged 2.0 mmol/l in both groups. Fenofibrate significantly reduced the incidence of both microalbuminuria and macroalbuminuria. However, in a valid pre-specified analysis of patients with triglycerides >2.3 mmol/l and HDL-cholesterol <0.9 mmol/l (approximately 17% of the ACCORD-Lipid population), combined therapy achieved an additional 31% reduction in cardiovascular risk, though this did not achieve statistical significance. ACCORD Lipid did not support the use of combined statin and fenofibrate therapy in the majority of T2D patients, but in those who had hypertriglyceridemia with or without low HDL-cholesterol, adding a fibrate was beneficial in reducing residual CVD risk, despite intensification of statin therapy.
(ii) ACCORD-Eye trial: In a subgroup of 2856 T2D patients, ACCORD-Eye investigated the effects of intensive glycemic control (glycated hemoglobin <6% vs. 7.0 to 7.9%), systolic blood pressure (<120 vs. <140 mmHg) and combination therapy for dyslipidemia (fenofibrate plus simvastatin vs. simvastatin alone) on retinopathy, assessed using fundal photography [101]. At 4 years, progression of retinopathy reduced significantly with improved control of glycemia (10.4% vs. 7.3%, p = 0.003), and dyslipidemia (10.2% vs. 6.5%, p = 0.006), but not with blood pressure control (8.8% vs. 10.4%, p = 0.29). Neither improved control of glycemia nor dyslipidemia had a significant effect on moderate vision loss. Progression of retinopathy with fenofibrate was apparently global and not confined to patients with diabetic dyslipidemia. The microvascular benefits of intensive glycemic control were counteracted by an increase in total and cardiovascular disease-related mortality, weight gain, and hypoglycemia [102]. That intensive blood pressure control did not reduce retinopathy progression may reflect the narrow range of blood pressure, small treatment effect, and short duration of the intervention [6, 101].
(iii) Implications of ACCORD: ACCORD-lipid supports recommendations that fibrates, in particular fenofibrate, may be used to treat residual dyslipidemia in statin-treated T2D patients [3, 4, 34]. Because ACCORD tended to recruit patients with low HDL-cholesterol, a conservative estimate of the proportion of T2D patients requiring the addition of fenofibrate is more likely to be ≤10%, especially in those treated with more potent statins [6]. Dyslipidemia is a risk factor for retinopathy [103], and, in the presence of both, the addition of fenofibrate to a statin may have value [6].
Statins and niacins. Combination therapy with a statin and niacin is associated with regression of coronary atherosclerosis and carotid intima-media thickness (CIMT) in patients at high cardiovascular risk with low HDL-cholesterol levels, including those with diabetes [104-107]. In the HDL-Atherosclerosis Treatment Study (HATS), combined statin and niacin therapy slowed the progression of coronary atherosclerosis in patients at high cardiovascular risk with low HDL-cholesterol levels, including those with diabetes [104]. The Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER 2) study, reported a significant inhibition in the progression of ultrasonographic CIMT in patients with known CAD and low levels of HDL-cholesterol, of whom 50% had the metabolic syndrome and 28% diabetes following 12 months of combination simvastatin and extended release (ER) niacin (Niaspan®) [105]. Patients who completed ARBITER 2 were subsequently enrolled in a follow-up study (ARBITER 3) [106]. In this follow-up study, a significant additional regression in CIMT was demonstrated over 24 months of treatment with ER niacin. ER niacin was also associated with a 23% increase in HDL-cholesterol, and this increase was independently associated with CIMT regression [106]. The ARBITER-6-HDL and LDL Treatment Strategies in Atherosclerosis (HALTS) study enrolled patients with CVD or CVD risk equivalent who were treated with long-term statin therapy to LDL-cholesterol levels <2.6 mmol/l, but who had residual low HDL-cholesterol levels. Patients were randomized to receive either ER niacin or ezetimibe [107]. The addition of ER niacin achieved a significant regression in CIMT compared with progression in CIMT with ezetimibe, despite a greater reduction in LDL-cholesterol with ezetimibe [107]. These beneficial effects may reflect the reduction in triglycerides and increase in HDL-cholesterol seen with ER niacin [6].
However, given the recent negative reports from the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL-cholesterol/High Triglyceride and Impact on Global Health Outcomes) study and the HPS2-THRIVE (Heart Protection Study-2 and the Treatment of HDL to reduce the incidence of vascular events), caution is indicated with the addition of niacin to statins in individuals with low HDL-cholesterol. AIM-HIGH was discontinued after 36 months of follow-up due to lack of clinical benefit from the addition of ER niacin to statin (± ezetimibe) therapy, despite significant improvements in HDL-cholesterol and triglycerides [108]. A further consideration in discontinuing AIM-HIGH was an increased stroke risk, but overall this was less than 1% and previous studies have not demonstrated an increased stroke risk with niacin.
A recent report from the AIM-HIGH trial suggests a non-significant trend towards benefits with the addition of ER niacin in a subset of patients in both the highest triglyceride and lowest HDL-cholesterol tertiles (p = 0.073) [109]. However, the investigators concluded that lipoprotein levels were not predictive of the differential benefit or harm with niacin treatment [109]. HPS2-THRIVE compared tredaptive (ER niacin 2 g plus laropiprant (an antiflushing agent) 40 mg daily) with placebo in 25, 673 patients treated with simvastatin plus ezetimibe if required. Following 3.9 years of follow-up, no significant benefit was demonstrated on the primary endpoint of major cardiovascular events, and a four-fold greater rate of myopathy was reported with tredaptive across all subgroups [110, 111]. Importantly, the safety data showed an excess of new onset diabetes, diabetic complications, infections, bleeding (gastrointestinal (GI) and intracranial), and GI symptoms with tredaptive. A caveat is that the volunteers were mainly Chinese and had very well-controlled LDL-cholesterol (1.6 mmol/l at baseline), so that the use of this agent, which has now been withdrawn from the market by Merck, should not be used in people with low HDL cholesterol/high triglyceride and LDL-cholesterol at target. A recent review suggests that the CV benefits of niacin alone may be independent of effects on HDL-cholesterol [112]. Whether any such benefits from niacin ER in HPS2-THRIVE were nullified by laropiprant, a prostaglandin D2 inhibitor, remains open to question.
Statins and n-3 fatty acids ethyl esters. Evidence suggests combined statin and n-3 fatty acids may reduce cardiovascular events in both a primary and secondary prevention setting. The Japan Eicosapentaenoic Acid Lipid Intervention Study (JELIS) demonstrated a reduction in major cardiovascular events with combined n-3 fatty acids (EPA 1800 mg daily) and low-dose statin (pravastatin 10 mg or simvastatin 5 mg daily) compared with statin alone [113]. However, this cardiovascular benefit may relate in part to the antiarrhythmic effects of n-3 fatty acids, and is independent of minor changes in plasma triglycerides [6, 20]. The ORIGIN trial found a lack of cardiovascular benefit with daily supplementation of 1 g of n-3 fatty acids (omacor) in patients with or at risk of T2D (50% treated with a statin) [93]. The benefit of adding higher-dose EPA to statins in hypertriglyceridemic subjects at high CVD risk is currently being tested in the REDUCE-IT trial (A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients with Hypertriglyceridemia and on Statin) [114].
6. Conclusions
Type 2 diabetic patients are at a markedly increased risk of CVD events. Dyslipidemia is a common risk factor and a strong predictor of CVD in T2D patients. Although statins decrease the incidence of CVD in T2D, residual cardiovascular risk remains high, despite the achievement of optimal or near-optimal plasma LDL cholesterol concentrations. This may, in part, be due to uncorrected atherogenic dyslipidemia. Therapeutic interventions, including lifestyle changes and lipid-regulating agents, correct diabetic dyslipidemia via several mechanisms. Recent evidence suggests that residual diabetic dyslipidemia and cardiovascular risk in statin-treated patients with T2D may be targeted with fenofibrate. At present, there are no clinical end-point trials supporting the addition of ezetimibe or marine-derived n-3 polyunsaturated fatty acids. In our opinion and on the basis of a recent clinical trial, niacin should not be added to a statin in individuals with low high-density lipoprotein cholesterol and very well controlled LDL-cholesterol.
Disclosures: GFW has received honoraria for Advisory Boards and lectures from the following industry related companies: Pfizer, Merck Sharpe and Dohme, Solvay, AstraZeneca, GalaxoSmithKline, Sanofi-Aventis.
References
- Turner RC, Millns H, Neil AW, Stratton IM. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom prospective diabetes study (UKPDS:23). Br Med J 1998. 316(7134):823-828. [DOD] [CrossRef]
- Fruchart JC, Sacks F, Hermans MP, Assmann G, Brown WV, Ceska R, Chapman MJ, Dodson PM, Fioretto P, Ginsberg HN, et al. The Residual Risk Reduction Initiative: A Call to Action to Reduce Residual Vascular Risk in Patients with Dyslipidemia. Am J Cardiol 2008. 102(10 Supplement 1):1K-34K. [DOD] [CrossRef]
- National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002. 106(25):3143-3421. [DOD]
- Brunzell Jdavidson MM, Furberg CM, Goldberg RM, Howard BP, Stein JM, Witztum JM. Lipoprotein Management in Patients With Cardiometabolic Risk: Consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008. 31(4):811. [DOD] [CrossRef]
- Taskinen R. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 2003. 46(6):733-749. [DOD] [CrossRef]
- Watts GF, Karpe F. Triglycerides and atherogenic dyslipidaemia: extending treatment beyond statins in the high-risk cardiovascular patient. Heart 2011. 97(5):350-356. [DOD] [CrossRef]
- Nordestgaard BG, Benn MM, Schnohr PM, Tybjaerg-Hansen AM. Nonfasting Triglycerides and Risk of Myocardial Infarction, Ischemic Heart Disease, and Death in Men and Women. JAMA 2007. 298(3):299-308. [DOD] [CrossRef]
- Sarwar N. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet 2010. 375(9726):1634-1639. [DOD] [CrossRef]
- Jorgensen AB, Frikke-Schmidt R, West AS, Grande P, Nordestgaard BG, Tybjaerg-Hansen A. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. Eur Heart J 2012. In press. [DOD]
- Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol 2013. 61(4):427-436. [DOD] [CrossRef]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012. 380(9841):572-580. [DOD] [CrossRef]
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 2012. 367(22):2089-2099. [DOD] [CrossRef]
- Syvanne M, Taskinen MR. Lipids and lipoproteins as coronary risk factors in non-insulin-dependent diabetes mellitus. Lancet 1997. 350(Suppl 1):SI20-SI23. [DOD] [CrossRef]
- Howard BV. Insulin resistance and lipid metabolism. Am J Cardiol 1999. 84(Supplement 1):28-32. [DOD] [CrossRef]
- Adiels M, Boren J, Caslake MJ, Stewart P, Soro A, Westerbacka J, Wennberg B, Olofsson SO, Packard C, Taskinen MR. Overproduction of VLDL1 Driven by Hyperglycemia Is a Dominant Feature of Diabetic Dyslipidemia. Arterioscler Thromb Vasc Biol 2005. 25(8):1697-1703. [DOD] [CrossRef]
- Adiels M, Taskinen MR, Packard C, Caslake M, Soro-Paavonen A, Westerbacka J, Vehkavaara S, Häkkinen A, Olofsson SO, Yki-Järvinen H, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006. 49(4):755-765. [DOD] [CrossRef]
- Cummings MH, Watts GF, Umpleby AM, Hennessy TR, Kelly JM, Jackson NC, Sönksen PH. Acute hyperinsulinemia decreases the hepatic secretion of very-low-density lipoprotein apolipoprotein B-100 in NIDDM. Diabetes 1995. 44(9):1059-1065. [DOD] [CrossRef]
- Malmstrom R, Packard CJ, Caslake M, Bedford D, Stewart P, Yki-Järvinen H, Shepherd J, Taskinen R. Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia 1997. V40(4):454-462. [DOD]
- Adiels M, Westerbacka J, Soro-Paavonen A, Häkkinen A, Vehkavaara S, Caslake M, Packard C, Olofsson S, Yki-Järvinen H, Taskinen M, et al. Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia 2007. 50(11):2356-2365. [DOD] [CrossRef]
- Chan DC, Watts GF. Dyslipidaemia in the metabolic syndrome and type 2 diabetes: pathogenesis, priorities, pharmacotherapies. Expert Opin Pharmacother 2010. 12(1):13-30. [DOD] [CrossRef]
- De Man FH, Castro Cabezas M, Van Barlingen HH, Erkelens DW, de Bruin TW. Triglyceride-rich lipoproteins in non-insulin-dependent diabetes mellitus: post-prandial metabolism and relation to premature atherosclerosis. Eur J Clin Invest 1996. 26(2):89-108. [DOD] [CrossRef]
- Woodman RJ, Chew GT, Watts GF. Mechanisms, significance and treatment of vascular dysfunction in type 2 diabetes mellitus: focus on lipid-regulating therapy. Drugs 2005. 65(1):31-74. [DOD] [CrossRef]
- Watts GF, Playford DA. Dyslipoproteinaemia and hyperoxidative stress in the pathogenesis of endothelial dysfunction in non-insulin dependent diabetes mellitus: a hypothesis. Atherosclerosis 1998. 141(1):17-30. [DOD] [CrossRef]
- Watts GF, Mandalia S, Brunt JN, Slavin BM, Coltart DJ, Lewis B. Independent associations between plasma lipoprotein subfraction levels and the course of coronary artery disease in the St. Thomas' Atherosclerosis Regression Study (STARS). Metabolism 1993. 42(11):1461-1467. [DOD] [CrossRef]
- Anber V, Griffin BA, McConnell M, Packard CJ, Shepherd J. Influence of plasma lipid and LDL-subfraction profile on the interaction between low density lipoprotein with human arterial wall proteoglycans. Atherosclerosis 1996. 124(2):261-271. [DOD] [CrossRef]
- Syvanne M, Kahri J, Virtanen KS, Taskinen MR. Coronary heart disease/myocardial infarction/systemic vascular responses: HDLs containing apolipoproteins A-I and A-II (LpA-I: A-II) as markers of coronary artery disease in men with non-insulin-dependent diabetes mellitus. Circulation 1995. 92(3):364-370. [DOD] [CrossRef]
- Syvanne M, Castro G, Dengremont C, De Geitere C, Jauhiainen M, Ehnholm C, Michelagnoli S, Franceschini G, Kahri J, Taskinen MR. Cholesterol efflux from Fu5AH hepatoma cells induced by plasma of subjects with or without coronary artery disease and non-insulin-dependent diabetes: importance of LpA-I:A-II particles and phospholipid transfer protein. Atherosclerosis 1996. 127(2):245-253. [DOD] [CrossRef]
- Nofer JR, Kehrel B, Fobker M, Levkau B, Assmann G, Eckardstein AV. HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis 2002. 161(1):1-16. [DOD] [CrossRef]
- Persegol L, Verges B, Foissac M, Gambert P, Duvillard L. Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 2006. 49(6):1380. [DOD] [CrossRef]
- Chapman MJ, Redfern JS, McGovern ME, Giral P. Niacin and fibrates in atherogenic dyslipidemia: Pharmacotherapy to reduce cardiovascular risk. Pharmacol Ther 2010. 126(3):314-345. [DOD] [CrossRef]
- American Diabetes Association. Standards of Medical Care in Diabetes-2013. Diabetes Care 2013. 36:S11-S66. [DOD] [CrossRef]
- Grundy SM, Cleeman JI, Merz CN, Brewer HB, Clark LT, Hunninghake DB, Pasternak RC, Smith SC, Stone NJ. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 2004. 44(3):720-732. [DOD] [CrossRef]
- National Vascular Disease Prevention Alliance. Guidelines for the management of absolute cardiovascular risk. 2012. [DOD]
- Graham I, Atar D, Borch-Johnsen K, Boysen G, Burell G, Cifkova R, Dallongeville J, De Backer G, Ebrahim S, Gjelsvik BR, et al. European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Eur Heart J 2007. 28(19):2375-2414. [DOD] [CrossRef]
- Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, et al. ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J 2011. 32(14):1769-1818. [DOD] [CrossRef]
- Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Boren J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J 2011. 32(11):1345-1361. [DOD] [CrossRef]
- Miller MM, Stone NJ, Ballantyne CM, Bittner VM, Criqui MH, Ginsberg HN, Goldberg AC, Howard WJ, Jacobson MS, et al. Triglycerides and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2011. 123(20):2292-2333. [DOD] [CrossRef]
- Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003. 361(9374):2005-2016. [DOD] [CrossRef]
- Colhoun PH, Betteridge PD, Durrington PP, Hitman PG, Neil HA, Livingstone SJ, Thomason MJ, Mackness MI, Charlton-Menys V, Fuller PJ. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004. 364(9435):685-696. [DOD] [CrossRef]
- Shepherd J, Barter P, Carmena R, Deedwania P, Fruchart JC, Haffner S, Hsia J, Breazna A, LaRosa J, Grundy S, Waters D. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the treating to new targets (TNT) study. Diabetes Care 2006. 29(6):1220-1226. [DOD] [CrossRef]
- Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Peto R, Armitage J, Baigent C. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008. 371(9607):117-125. [DOD] [CrossRef]
- Ginsberg H. Effects of Combination Lipid Therapy in Type 2 Diabetes Mellitus. N Engl J Med 2010. 362(17):1563-1574. [DOD] [CrossRef]
- Chan DC, Watts GF, Ng TW, Yamashita S, Barrett PH. Effect of weight loss on markers of triglyceride-rich lipoprotein metabolism in the metabolic syndrome. Eur J Clin Invest 2008. 38(10):743-751. [DOD] [CrossRef]
- Ng TW, Watts GF, Barrett PH, Rye KA, Chan DC. Effect of weight loss on LDL and HDL kinetics in the metabolic syndrome: associations with changes in plasma retinol-binding protein-4 and adiponectin levels. Diabetes Care 2007. 30(11):2945-2950. [DOD] [CrossRef]
- Watts GF, Chan DC. Mechanisms for therapeutic correction of dyslipidaemia in insulin resistance and diabetes. Atheroscler Suppl 2010. 11(1):61-64. [DOD] [CrossRef]
- Fujioka K. Benefits of moderate weight loss in patients with type 2 diabetes. Diabetes Obes Metab 2010. 12(3):186-194. [DOD] [CrossRef]
- Pi-Sunyer X, Blackburn G, Brancati F, Bray G, Bright R, Clark JM, Curtis JM, Espeland MA, Foreyt JP, Graves K, Haffner SM, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes. Diabetes Care 2007. 30(6):1374-1383. [DOD] [CrossRef]
- Wing R. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: four-year results of the Look AHEAD Trial. Arch Intern Med 2010. 170(17):1566-1575. [DOD]
- Alam S, Stolinski M, Pentecost C, Boroujerdi MA, Jones RH, Sonksen PH, Umpleby AM. The Effect of a six-month exercise program on very low-density lipoprotein apolipoprotein B secretion in type 2 diabetes. J Clin Endocrinol Metab 2004. 89(2):688-694. [DOD] [CrossRef]
- Deedwania P, Barter P, Carmena R, Fruchart JC, Grundy SM, Haffner S, Kastelein JJ, LaRosa JC, Schachner H, Shepherd J, et al. Reduction of low-density lipoprotein cholesterol in patients with coronary heart disease and metabolic syndrome: analysis of the Treating to New Targets study. Lancet 2006. 368(9539):919-928. [DOD] [CrossRef]
- The Diabetes Atorvastin Lipid Intervention Study Group. The effect of aggressive versus standard lipid lowering by atovastatin on diabetic dyslipidemia: The DALI Study: A double-blind, randomized, placebo-controlled trial in. Diabetes Care 2001. 24(8):1335. [DOD] [CrossRef]
- Isley WL, Harris WS, Miles JM. The effect of high-dose simvastatin on free fatty acid metabolism in patients with type 2 diabetes mellitus. Metabolism 2006. 55(6):758-762. [DOD] [CrossRef]
- Pontrelli L, Parris W, Adeli K, Cheung RC. Atorvastatin treatment beneficially alters the lipoprotein profile and increases low-density lipoprotein particle diameter in patients with combined dyslipidemia and impaired fasting glucose/type 2 diabetes. Metabolism 2002. 51(3):334-342. [DOD] [CrossRef]
- Stein EA, Lane M, Laskarzewski P. Comparison of Statins in Hypertriglyceridemia. Am J Cardiol 1998. 81(Suppl 1):66B-69B. [DOD] [CrossRef]
- Stein E, Plotkin D, Bays H, Davidson M, Dujovne C, Korenman S, Stepanavage M, Mercuri M. Effects of simvastatin (40 and 80 mg/day) in patients with mixed hyperlipidemia. Am J Cardiol 2000. 86(4):406-411. [DOD] [CrossRef]
- Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E. Impact of Triglyceride Levels Beyond Low-Density Lipoprotein Cholesterol After Acute Coronary Syndrome in the PROVE IT-TIMI 22 Trial. J Am Coll Cardiol 2008. 51(7):724-730. [DOD] [CrossRef]
- Verges B, Florentin E, Baillot-rudoni S, Monier S, Petit J, Rageot D, Gambert P, Duvillard L. Effects of 20 mg rosuvastatin on VLDL1-, VLDL2-, IDL- and LDL-ApoB kinetics in type 2 diabetes. Diabetologia 2008. 51(8):1382-1390. [DOD] [CrossRef]
- Watts GF, Barrett PH, Ji J, Serone AP, Chan DC, Croft KD, Loehrer F, Johnson AG. Differential regulation of lipoprotein kinetics by atorvastatin and fenofibrate in subjects with the metabolic syndrome. Diabetes 2003. 52(3):803-811. [DOD] [CrossRef]
- Chan DC, Watts GF, Barrett PH, Beilin LJ, Redgrave TG, Mori TA. Regulatory Effects of HMG CoA Reductase Inhibitor and Fish Oils on Apolipoprotein B-100 Kinetics in Insulin-Resistant Obese Male Subjects With Dyslipidemia. Diabetes 2002. 51(8):2377-2386. [DOD] [CrossRef]
- Shepherd J, Barter P, Carmena R, Deedwania P, Fruchart JC, Haffner S, Hsia J, Breazna A, LaRosa J, Grundy S, et al. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006. 29(6):1220-1226. [DOD] [CrossRef]
- Sever PS, Poulter NR, Dahlof B, Wedel H, Collins R, Beevers G, Caulfield M, Kjeldsen SE, Kristinsson A, McInnes GT, et al. Reduction in cardiovascular events with atorvastatin in 2,532 patients with type 2 diabetes: Anglo-Scandinavian Cardiac Outcomes Trial--lipid-lowering arm (ASCOT-LLA). Diabetes Care 2005. 28(5):1151-1157. [DOD] [CrossRef]
- Jun M, Foote C, Lv J, Neal B, Patel A, Nicholls SJ, Grobbee DE, Cass A, Chalmers J, Perkovic V. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet 2010. 375(9729):1875-1884. [DOD] [CrossRef]
- Jun M, Zhu B, Tonelli M, Jardine MJ, Patel A, Neal B, Liyanage T, Keech A, Cass A, Perkovic V. Effects of fibrates in kidney disease: a systematic review and meta-analysis. J Am Coll Cardiol 2012. 60(20):2061-2071. [DOD] [CrossRef]
- Gervois P, Fruchart JC, Staels B. Drug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nat Clin Pract Endocrinol Metab 2007. 3(2):145-156. [DOD]
- Koskinen P, Manttari M, Manninen V, Huttunen JK, Heinonen OP, Frick MH. Coronary heart disease incidence in NIDDM patients in the Helsinki Heart Study. Diabetes Care 1992. 15(7):820-825. [DOD] [CrossRef]
- Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, Forder P. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005. 366(9500):1849-1861. [DOD] [CrossRef]
- Scott R, O'Brien R, Fulcher G, Pardy C, d'Emden M, Tse DP, Taskinen MM, Ehnholm CM, Keech A. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome. Diabetes Care 2009. 32(3):493. [DOD] [CrossRef]
- Davis T, Ting R, Best J, Donoghoe M, Drury P, Sullivan D, Jenkins A, O'Connell R, Whiting M, Glasziou P, et al. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia 2011. 54(2):280. [DOD] [CrossRef]
- Keech AC, Mitchell P, Summanen PA, O'Day J, Davis TME, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet 2007. 370(9600):1687-1697. [DOD] [CrossRef]
- Rajamani K, Colman PG, Li LP, Best JD, Voysey M, D’Emden MC, Laakso M, Baker JR, Keech AC. Effect of fenofibrate on amputation events in people with type 2 diabetes mellitus (FIELD study): a prespecified analysis of a randomised controlled trial. Lancet 2009. 373(9677):1780-1788. [DOD] [CrossRef]
- Shepherd J, Betteridge J, Van Gaal L. Nicotinic acid in the management of dyslipidaemia associated with diabetes and metabolic syndrome: a position paper developed by a European Consensus Panel. Curr Med Res Opin. 2005. 21(5):665-682. [DOD]
- Karpe F, Frayn KN. The nicotinic acid receptor-a new mechanism for an old drug. Lancet 2004. 363(9424):1892-1894. [DOD] [CrossRef]
- Vosper H. Niacin: a re-emerging pharmaceutical for the treatment of dyslipidaemia. Br J Pharmacol 2009. 158:429-441. [DOD] [CrossRef]
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 2003. 9(3):352-355. [DOD] [CrossRef]
- Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, Ignar DM, Murdock PR, Steplewski K, Green A, et al. Molecular Identification of High and Low Affinity Receptors for Nicotinic Acid. J Biol Chem 2003. 278(11):9869-9874. [DOD] [CrossRef]
- Soga T, Kamohara M, Takasaki J, Matsumoto Si, Saito T, Ohishi T, Hiyama H, Matsuo A, Matsushime H, Furuichi K. Molecular identification of nicotinic acid receptor. Biochem Biophys Res Commun 2003. 303(1):364-369. [DOD] [CrossRef]
- Tunaru S, Lattig J, Kero J, Krause G, Offermanns S. Characterization of Determinants of Ligand Binding to the Nicotinic Acid Receptor GPR109A (HM74A/PUMA-G). Mol Pharmacol 2005. 68(5):1271-1280. [DOD] [CrossRef]
- van der Hoorn JW, de Haan W, Berbee JF, Havekes LM, Jukema JW, Rensen PC, Princen HM. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol 2008. 28(11):2016-2022. [DOD] [CrossRef]
- Ganji SH, Tavintharan S, Zhu D, Xing Y, Kamanna VS, Kashyap ML. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J Lipid Res 2004. 45(10):1835-1845. [DOD] [CrossRef]
- Guyton JR. Extended-release niacin for modifying the lipoprotein profile. Expert Opin Pharmacother 2004. 5(6):1385-1398. [DOD] [CrossRef]
- Kamanna VS, Kashyap ML. Mechanism of action of niacin on lipoprotein metabolism. Curr Atheroscler Rep 2000. 2(1):36-46. [DOD] [CrossRef]
- Lamon-Fava S, Diffenderfer MR, Barrett PH, Buchsbaum A, Nyaku M, Horvath KV, Asztalos BF, Otokozawa S, Ai M, Matthan NR, et al. Extended-Release Niacin Alters the Metabolism of Plasma Apolipoprotein (Apo) A-I and ApoB-Containing Lipoproteins. Arterioscler Thromb Vasc Biol 2008. 28(9):1672-1678. [DOD] [CrossRef]
- Berge KG, Canner PL. Coronary drug project: Experience with niacin. Eur J Clin Pharmacol 1991. 40(1):S49-S51. [DOD] [CrossRef]
- Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald W. Fifteen year mortality in Coronary Drug Project patients: Long-term benefit with niacin. J Am Coll Cardiol 1986. 8(6):1245-1255. [DOD] [CrossRef]
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005. 95(2):254-257. [DOD] [CrossRef]
- Canner PL, Furberg CD, McGovern ME. Benefits of Niacin in Patients With Versus Without the Metabolic Syndrome and Healed Myocardial Infarction (from the Coronary Drug Project). Am J Cardiol 2006. 97(4):477-479. [DOD] [CrossRef]
- Guyton JR, Bays HE. Safety considerations with niacin therapy. Am J Cardiol 2007. 99(6A):22C-31C. [DOD] [CrossRef]
- Grundy SM, Vega GL, McGovern ME, Tulloch BR, Kendall DM, Fitz-Patrick D, Ganda OP, Rosenson RS, Buse JB, Robertson DD, et al. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the assessment of diabetes control and evaluation of the efficacy of niaspan trial. Arch Intern Med 2002. 162(14):1568-1576. [DOD]
- Goldberg RB, Jacobson TA. Effects of niacin on glucose control in patients with dyslipidemia. Mayo Clin Proc 2008. 83(4):470-478. [DOD] [CrossRef]
- Jump DB. N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr Opin Lipidol 2008. 19(3):242-247. [DOD] [CrossRef]
- Durrington PN, Bhatnagar D, Mackness MI, Morgan J, Julier K, Khan MA, France M. An omega-3 polyunsaturated fatty acid concentrate administered for one year decreased triglycerides in simvastatin treated patients with coronary heart disease and persisting hypertriglyceridaemia. Heart 2001. 85(5):544-548. [DOD] [CrossRef]
- GISSI-Prevenzionne Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet 1999. 354(9177):447-455. [DOD] [CrossRef]
- Bosch J, Gerstein HC, Dagenais GR, Diaz R, Dyal L, Jung H, Maggiono AP, Probstfield J, Ramachandran A, Riddle MC, Ryden LE, Yusuf S. N-3 Fatty Acids and Cardiovascular Outcomes in Patients with Dysglycemia. N Engl J Med 2012. 367(4):309-318. [DOD] [CrossRef]
- Saravanan P, Davidson NC, Schmidt EB, Calder PC. Cardiovascular effects of marine omega-3 fatty acids. Lancet 2010. 376(9740):540-550. [DOD] [CrossRef]
- Faergeman O, Holme I, Fayyad R, Bhatia S, Grundy SM, Kastelein JJ, LaRosa JC, Larsen ML, Lindahl C, Olsson AG, et al. Plasma Triglycerides and Cardiovascular Events in the Treating to New Targets and Incremental Decrease in End-Points Through Aggressive Lipid Lowering Trials of Statins in Patients With Coronary Artery Disease. Am J Cardiol 2009. 104(4):459-463. [DOD] [CrossRef]
- Bottorff MB. Statin Safety and Drug Interactions: Clinical Implications. Am J Cardiol 2006. 97(Suppl 1):S27-S31. [DOD] [CrossRef]
- Pandor A, Ara RM, Tumur I, Wilkinson AJ, Paisley S, Duenas A, Durrington PN, Chilcott J. Ezetimibe monotherapy for cholesterol lowering in 2,722 people: systematic review and meta-analysis of randomized controlled trials. J Intern Med 2009. 265(5):568-580. [DOD] [CrossRef]
- Goldberg RB, Guyton JR, Mazzone T, Weinstock RS, Polis A, Edwards P, Tomassini JE, Tershakovec AM. Ezetimibe/simvastatin vs atorvastatin in patients with type 2 diabetes mellitus and hypercholesterolemia: the VYTAL study. Mayo Clin Proc 2006. 81(12):1579-1588. [DOD] [CrossRef]
- Fleg JL, Mete M, Howard BV, Umans JG, Roman MJ, Ratner RE, Silverman A, Galloway JM, Henderson JA, Weir MR, et al. Effect of statins alone versus statins plus ezetimibe on carotid atherosclerosis in type 2 diabetes: the SANDS (Stop Atherosclerosis in Native Diabetics Study) Trial. J Am Coll Cardiol 2008. 52(25):2198-2205. [DOD] [CrossRef]
- Roth EM, Rosenson RS, Jones PH, Davidson MH, Kelly MT, Setze CM, Lele A, Thakker K. Attainment of goal/desirable lipid levels in patients with mixed dyslipidemia after 12 weeks of treatment with fenofibric acid and rosuvastatin combination therapy: A pooled analysis of controlled studies. J Clin Lipidol 2012. 6(6):534-544. [DOD] [CrossRef]
- Chew E, Ambrosius W, Davis M, Danis R, Gangaputra S, Greven C, et al. Effects of Medical Therapies on Retinopathy Progression in Type 2 Diabetes. N Engl J Med 2010. 363(3):233-244. [DOD] [CrossRef]
- Ismail-Beigi F, Craven T, Banerji MA, Basile J, Calles J, Cohen RM, Cuddihy R, Cushman WC, Genuth S, Grimm RH Jr, et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010. 376(9739):419-430. [DOD] [CrossRef]
- Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet 2010. 376(9735):124-136. [DOD] [CrossRef]
- Brown BG, Xue-Qiao Z, Alan C, Lloyd DF. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med 2001. 345(22):1583. [DOD] [CrossRef]
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: A Double-Blind, Placebo-Controlled Study of Extended-Release Niacin on Atherosclerosis Progression in Secondary Prevention Patients Treated With Statins. Circulation 2004. 110(23):3512-3517. [DOD] [CrossRef]
- Taylor A, Lee H, Sullenberger L. The effect of 24 months of combination statin and extended-release niacin on carotid intima-media thickness: ARBITER 3*. Curr Med Res Opin 2006. 22(11):2243-2250. [DOD] [CrossRef]
- Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M, Weissman NJ, Turco M. Extended-Release Niacin or Ezetimibe and Carotid Intima-Media Thickness. N Engl J Med 2009. 361(22):2113-2122. [DOD] [CrossRef]
- Boden WE, Probstfield JK, Anderson T, Chaitman BR, Koprowicz K, Teo K. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N Engl J Med 2011. 365(24):2255-2267. [DOD] [CrossRef]
- Guyton JR, Slee AE, Anderson T, Fleg JL, Goldberg RB, Kashyap ML, Marcovina SM, Nassh SD, O’Brien KD, Weomtraib W, et al. Abstract 14501: Relation of Lipoprotein Levels to Cardiovascular Events in the Atherotrhombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes (AIM_HIGH) Trail. Circulation 2012. 126(21):A14501. [DOD]
- HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 2013. 34:1279-1291. [DOD] [CrossRef]
- HPS-2 THRIVE misses primary end point: No benefit of niacin/laropiprant. http://www.theheart.org/article/1490635.do. [DOD]
- Lavigne PM, Karas RH. The current state of niacin in cardiovascular disease prevention: a systematic review and meta-regression. J Am Coll Cardiol 2013. 61(4):440-446. [DOD] [CrossRef]
- Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura H, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007. 369(9567):1090-1098. [DOD] [CrossRef]
- Clinical Trials.gov. A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients With Hypertriglyceridemia and on Statin (REDUCE-IT). http://clinicaltrials.gov/show/NCT01492361. [DOD]
This article has been cited by other articles:

|
Dyslipidemia in diabetes mellitus and cardiovascular disease
Warraich HJ, Rana JS
Cardiovasc Endocrinol 2017. 6(1):27-32
|
|

|
The Atherogenic Dyslipidemia Ratio Log (Tg)/Hdl-C Was Not Associated with Urinary Albumin Excretion Rate (Uaer) and Increased Cardiovascular Risk in Black Patients with Type 2 Diabetes
Kajingulu MF, Lepira BF, Mbutiwi IN, Makulo JR, Bieleli E, Nseka MN
World J Cardiovasc Dis 2016. 6(1):14-20
|
|

|
Role for combination therapy in diabetic dyslipidemia
Warraich HJ, Wong ND, Rana JS
Curr Cardiol Rep 2015. 17(5):589
|
|
|
Targeting metabolic disorders by natural products
Tabatabaei-Malazy O, Larijani B, Abdollahi M
J Diabetes Metab Disord 2015. 14:57
|
|

|
Diabetes Mellitus, Cognitive Impairment, and Traditional Chinese Medicine
Seto SW, Yang GY, Kiat H, Bensoussan A, Kwan YW, Chang D
Int J Endocrinol 2015. 2015:810439
|
|
|
Origin and therapy for hypertriglyceridaemia in type 2 diabetes
Pang J, Chan DC, Watts GF
World J Diabetes 2014. 5(2):165-175
|
|
|


)

