Chapter I. Pathogenesis and Function
| Rev Diabet Stud,
2015,
12(3-4):243-259 |
DOI 10.1900/RDS.2015.12.243 |
Novel Genetic Actors of Diabetes-Associated Microvascular Complications: Retinopathy, Kidney Disease and Neuropathy
Samaneh Davoudi, Lucia Sobrin
Massachusetts Eye and Ear Infirmary, Harvard Medical School, 243 Charles Street, Boston, MA 02114, USA
Address correspondence to: Lucia Sobrin, e-mail: lucia_sobrin@meei.harvard.edu
Manuscript submitted April 7, 2015; resubmitted April 14, 2015; accepted April 22, 2015.
Keywords: type 2 diabetes, GWAS, retinopathy, nephropathy, neuropathy, microvascular complications, HbA1c, SNP, linkage disequilibrium
Abstract
Both type 1 and type 2 diabetes mellitus can lead to the common microvascular complications of diabetic retinopathy, kidney disease, and neuropathy. Diabetic patients do not universally develop these complications. Long duration of diabetes and poor glycemic control explain a lot of the variability in the development of microvascular complications, but not all. Genetic factors account for some of the remaining variability because of the heritability and familial clustering of these complications. There have been a large number of investigations, including linkage studies, candidate gene studies, and genome-wide association studies, all of which have sought to identify the specific variants that increase susceptibility. For retinopathy, several genome-wide association studies have been performed in small or midsize samples, but no reproducible loci across the studies have been identified. For diabetic kidney disease, genome-wide association studies in larger samples have been performed, and loci for this complication are beginning to emerge. However, validation of the existing discoveries, and further novel discoveries in larger samples is ongoing. The amount of genetic research into diabetic neuropathy has been very limited, and much is dedicated to the understanding of genetic risk factors only. Collaborations that pool samples and aim to detect phenotype classifications more precisely are promising avenues for a better explanation of the genetics of diabetic microvascular complications.
Abbreviations: AER – albumin excretion rate; ALR – aldose reductase; CARe – Candidate Gene Association Resource; CI – confidence interval; CARS – cysteinyl-tRNA synthetase 9; DCCT – Diabetes Control and Complications Trial; DKD – diabetic kidney disease; DR – diabetic retinopathy; DME – diabetic macular edema; DNCRI – Diabetic Nephropathy Collaborative Research Initiative; DNAme – DNA methylation; EDS – Ealing Diabetes Study; eNOS – endothelial nitric oxide synthase; ESRD – end-stage renal disease; ELMO1 - engulfment and cell motility 1 gene; EDIC – Epidemiology of Diabetes Interventions and Complications Trial; EPO – erythropoietin; eGFR – estimated glomerular filtration rate; EURAGEDIC – European Rational Approach for the Genetics of Diabetic Complications; FIND – Family Investigation of Nephropathy and Diabetes; FinnDiane – Finnish Diabetic Nephropathy; GoDARTS – Genetics of Diabetes Audit and Research Tayside; GoKinD – Genetics of Kidneys in Diabetes; GENIE – Genetics of Nephropathy–an International Effort; GFRA2 – glial cell line derived neurotropic factor family receptor alpha 2; GPx-1 – glutathione peroxidase-1; HbA1c – glycated hemoglobin; GWAS – genome-wide association studies; IDUA – Iduronidase; miRNA – microRNA; NOS3 – nitric oxide synthase; NPDR – non-proliferative diabetic retinopathy; OR – odds ratio; PTM – post-translational modifications; PDR – proliferative diabetic retinopathy; PKC – protein kinase C; SELP – P selectin; RAGE – receptor for advanced glycation end-products; SNP – single nucleotide polymorphism; SUMMIT – Surrogate markers for Micro- and Macrovascular hard endpoints for Innovative diabetes Tools; TCF7L2 – transcription factor 7-like 2; TGF-β1 – transforming growth factor-beta 1; T1D – type 1 diabetes; T2D – type 2 diabetes; UK-ROI – United Kingdom-Republic of Ireland; UDACS – University College London Diabetes and Cardiovascular disease Study
1. Introduction
The typical microvascular complications of type 2 diabetes (T2D) are diabetic retinopathy, nephropathy, and neuropathy. They are responsible for a large portion of the morbidity associated with T2D, including blindness, end-stage renal disease, and extremity neuralgias [1]. The incidence and prevalence of microvascular complications strongly increase with duration of diabetes and glycated hemoglobin (HbA1c) [2-5]. The cells damaged by hyperglycemia include capillary endothelial cells in the retina, mesangial cells in the glomerulus, and Schwann cells and neurons in peripheral nerves [6, 7]. However, vascular complications can also occur in patients with HbA1c levels of less than 7% and even in undiagnosed patients because of transient elevations in serum glucose [3]. In the Diabetes Control and Complications Trial (DCCT), duration of diabetes and HbA1c explained only about 11% of the variation in the risk of retinopathy [8, 9].
Genetic variation is postulated to explain some of the remaining heterogeneity in diabetic microvascular complications. Twin and familial studies have documented clear familial clustering [10, 11]. In an attempt to find the genetic risk factors for microvascular complications, linkage studies, candidate gene association studies, and genome-wide association studies (GWAS) have been performed, and are currently being performed. They aim to shed some light on the genetic susceptibility for these complications. This review summarizes the findings of these studies into genetic risk factors for diabetic retinopathy, kidney disease, and neuropathy.
These complications also occur in patients with type 1 diabetes (T1D). It is still unknown whether the genes for microvascular complications overlap between the two types of diabetes, but it is likely that there is some if not considerable overlap. Many of the genetic studies for microvascular complications have been performed in mixed T1D and T2D populations. Although this review is dedicated to T2D, we also describe results for studies examining microvascular complications in both types of diabetes.
2. Diabetic retinopathy
2.1 Heritability and linkage studies
The initial evidence for diabetic retinopathy (DR) heritability came from twin studies. The concordance rates for the presence and severity of DR was higher in monozygotic than in the dizygotic twins [12]. Interestingly, there is a higher concordance of DR in T2D monozygotic twins (95%) than in T1D monozygotic twins (68%), suggesting that in T1D the initiation and development of its complications are less dependent on genetic factors than in T2D [11]. Additional family studies have shown that, depending on the DR phenotype and the ethnic population examined, siblings and relatives of diabetic patients with DR have approximately a 2- to 4-fold risk of developing the complication compared with relatives of diabetic patients without DR complication [5, 13-18]. The degree of familial aggregation is greater for more severe forms of retinopathy. Heritability has been estimated to be as high as 27% for DR and 52% for proliferative diabetic retinopathy (PDR), a more advanced form of the disease [13, 14].
Linkage studies have provided limited and inconsistent information about the potential genetic loci for DR. A sibling-pair linkage analysis for DR in Pima Indians with T2D found only modest evidence of linkage at chromosomes 3 and 9 with (logarithm of odds) LOD scores of 1.36 and 1.46, respectively [19]. A subsequent genome-wide linkage analysis in this population found stronger evidence for linkage on chromosome 1p with LOD scores of 2.58 and 3.1 for single-point and multi-point analyses, respectively [14]. Another genome-wide linkage scan for genes contributing to DR, using 794 diabetes participants from 393 Mexican-American families in Starr County, Texas, having at least two diabetic siblings, revealed only suggestive evidence of linkage with retinopathy on chromosomes 3 and 12 [20].
2.2 Candidate gene association studies
Candidate genes that have a putative role in DR pathophysiology have been studied by many groups. However, the results from candidate gene studies have been inconsistently replicated, as summarized in other articles [21-23]. Here, we will highlight some of the best powered candidate gene studies, and the more extensively examined candidate genes, including vascular endothelial growth factor (VEGF), erythropoietin (EPO), transcription factor 7-like 2 (TCF7L2), aldose reductase (ALR), receptor for advanced glycation end-products (RAGE) genes, and genes in the renin-angiotensin system [22].
The critical role of VEGF in the pathophysiology of PDR and diabetic macular edema (DME) is well-known. Anti-VEGF medications are the standard care for these advanced forms of DR [24]. Many different single nucleotide polymorphisms (SNPs) in VEGF genes have been examined, but there has been no consistently reproducible association. The VEGF C-634-G polymorphism (rs2010963) was one of the initially examined SNPs, and it was reported to be associated with DME and DR [25]. In 2007, the DCCT examined a panel of 18 VEGFA SNPs, all of which represent linkage disequilibrium bins (pairwise r2 = 0.64); it was found that they were collectively associated with severe retinopathy (p = 6.8 x 10-5) [26]. However, the rs2010963 polymorphism was not associated to a statistically significant degree in this trial. The most significant single SNP association was with rs3025021, p = 0.0017. Another analysis of VEGF SNPs on the risk of retinopathy was performed in 1,336 cases and 1,231 controls from non-Hispanic white T2D populations [27]. Despite this significant statistical power, the investigators did not find any significant effects of the two examined SNPs, rs6921438 or rs10738760, on retinopathy. Neither of these latter SNPs were in strong linkage disequilibrium with the SNPs investigated in the aforementioned studies. In a recent meta-analysis of eleven studies, no significant association was found for rs2010963; rs3025021, rs6921438, and rs10738760 were not examined. Links between DR and VEGF +936C/T (rs3025039, p = 0.01) and VEGF-460T/C (rs833061, p = 0.02) were detected by using recessive models [28].
Most studies have focused on VEGFA, but recently a genetic association study of SNPs in VEGFC and DR was performed [29]. Thirteen VEGFC tag SNPs were genotyped in 2,899 white patients with T1D and T2D. Participants with diabetes but not DR (n = 980) were compared with patients with diabetes and "any DR" (n = 1,919). Three VEGFC SNPs were associated with DR: rs17697419 (p = 0.001; OR: 0.67; CI: 0.52-0.85), rs17697515 (p = 0.001; OR: 0.62; CI: 0.47-0.81), and rs2333526 (p = 0.005; OR: 0.69; CI: 0.54-0.90). It appeared that rs17697515 was associated with DME in those with T2D (p = 0.004; OR: 0.53; CI: 0.35-0.82).
Another angiogenic factor, erythropoietin, has also been the focus of candidate gene studies. An association between the T allele of rs1617640 in the EPO promoter and PDR was first reported in 2008 [30]. The advantages of this study include:
- Large sample size (n = 2572 across three cohorts)
- Examination of the DR phenotype with greatest heritability (PDR)
- Stringent definition of controls as participants with diabetes for at least 15 years, but without retinopathy
- Consistency in the effect found across separate cohorts
A second, albeit smaller, study found the opposite allele of this same SNP in EPO to be associated with DR risk [31]. This EPO association also could not be replicated in the DCCT/EDIC (Epidemiology of Diabetes Interventions and Complications Trial) for DR or time to severe DR [32].
The TCF7L2 polymorphism rs7903146 is the common variant with strongest effect for T2D. An association with PDR in Caucasians with T2D and replication in an independent cohort was reported [33]. This study had a reasonable sample size (n = 1,139 in the discovery and replication cohorts combined), used the PDR phenotype, and defined controls stringently with a minimum diabetes duration of 15 years. TCF7L2 had already been studied in DR with both positive [33, 34] and negative results [32, 35]. Further investigation in other T2D populations is warranted.
Abhary et al. performed a meta-analysis of 34 variants in twenty genes which were previously reported to be genetic risk factors for DR [22]. Data on the insertion/deletion polymorphism in intron 16 of the ACE gene were analyzed from six studies of patients with T1D and seven studies of patients with T2D. The 287 base pair deletion was treated as the risk variant, and there was no statistically significant association with this polymorphism and the development of any form of DR.
AKR1B1 is part of the aldose reductase pathway. The three most commonly investigated variants in the AKR1B1 (CA)n microsatellite (z, z+2, and z-2) were analyzed using results from six studies in T1D patients and nine studies in T2D patients. There was a significant association with the z-2 allele and the development of any DR (OR: 2.33; 95% CI: 1.49-3.64; p = 2 × 10-4). Subanalysis revealed a significant association between the z-2 allele and DR in patients with T2D (OR: 2.64, 95% CI: 1.39-5.01; p = 2.9 × 10-3), with a weaker but still statistically significant association for patients with T1D (OR: 1.95; 95% CI: 1.04-3.66; p = 0.04) [22].
RAGE is mainly an intracellular signal-transducer or pro-inflammatory peptide that may be of importance in inflammation and autoimmune diseases [1, 36]. The -374A T/A polymorphism influences a transcription factor-binding site which, in turn, leads to the upregulation of RAGE transcription. Some studies have found an association between the -374 T/A polymorphism and sight-threatening DR [37, 38]. Two meta-analysis performed in Caucasian and Asian populations, however, did not find a significant association between RAGE polymorphisms (-374A, Gly82Ser, and -429T/C) and DR [39, 40].
The Candidate Gene Association Resource (CARe) performed a candidate gene study for DR with a sample size larger than those previously used (n = 8,040, including discovery and replication cohorts) and comprehensive coverage of variations in 2,000 genes associated with cardiovascular, metabolic, and inflammatory pathways [41]. By not choosing specific genes, but rather examining pathways, and appropriately correcting for multiple-hypotheses testing, the investigation was less reliant on the accuracy of a priori hypotheses regarding the causal genes. In the discovery cohort, variants in P selectin (SELP) and iduronidase (IDUA) were significantly associated with DR after correction for multiple hypotheses, but these associations could not be replicated in independent cohorts. Of note, the study detected an association between EPO and DR consistent with the previously reported association [30], but the p-value was just below the threshold for significance after correction for multiple hypotheses. In 2015, Penman et al. performed a study on 629 patients with T2D [42]. In multivariate models, higher P-selectin levels were associated with any DR and PDR, and minor allele homozygotes for the SELP variant rs6128 were less likely to develop DR. The association between rs6128 and retinopathy was consistent, in terms of direction of effect, with that reported in CARe.
2.3 Genome-wide association studies
The results of GWAS performed for DR are summarized in Table 1. Like candidate gene studies, GWAS have not yet detected associated loci consistently. The first GWAS for DR was performed in a Mexican-American T2D population on an early genome-wide genotyping platform which covered common human variations less accurately than the more recent versions of the technology [43]. None of the SNPs examined met the threshold for genome-wide significance when 183 controls with no DR or early non-proliferative diabetic retinopathy (NPDR) were compared with 103 cases with moderate to severe NPDR or PDR. The phenotyping was based on the Early Treatment of Diabetic Retinopathy Study (ETDRS) grading of fundus photographs, in which the cases without DR and early NPDR were graded 10-37 and those with moderate NPDR to PDR graded 43-85. The second GWAS was executed in Genetics of Kidneys in Diabetes (GoKinD) and EDIC, a Caucasian T1D population with 973 severe retinopathy cases, defined by a questionnaire history of DR laser treatment, and 1,856 controls who had never received laser treatment [44]. Severe retinopathy cases were identified according to patient-reported history of prior laser photocoagulation for DR. Controls were all the remaining subjects in the cohort. This study did not identify any loci that were genome-wide significant [45]. The study found candidate loci which were subsequently examined in an independent sample. However, none of these loci achieved genome-wide significance in the subsequent analysis.
Table
1.
Top signals from genome-wide associations studies examining diabetic retinopathy |
|
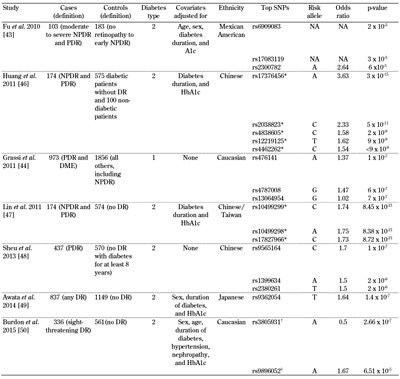 |
 |
Legend:
* These SNPs with p-values less than 5 x 10-8 have not been independently replicated by any study to date. † No association was found after replication. ‡ The p-value for this SNP is the result of a meta-analysis with independent replication cohorts. Abbreviations: DME – diabetic macular edema, DR – diabetic retinopathy, NA – not available, NPDR – non-proliferative diabetic retinopathy, PDR – proliferative diabetic retinopathy. SNP – single nucleotide polymorphism. |
|
A third GWAS for DR in a Chinese T2D cohort was performed with 174 NPDR and PDR cases and 675 controls with no DR as graded by an ophthalmologist according to the American Academy of Ophthalmology's proposed international scale for severity of clinical DR [46]. This investigation reported several variants associated with genome-wide significant p-values. However, it examined multiple genetic models, and did not correct for these additional hypotheses. Also, no independent replication was attempted. With appropriate correction for the number of models examined, the genome-wide significance would not remain. A fourth GWAS for DR in a Chinese T2D cohort with 102 NPDR and 42 PDR cases and 575 controls without DR reported novel DR risk loci on chromosome 6, which reached the generally accepted p-value for genome-wide statistical significance of less than 5 x 10-8 [47]. Fundus photographs were graded by ophthalmologists according to the international scale for severity of clinical DR set forth by the American Academy of Ophthalmology. Again, six models were examined, but these additional models were not considered in multiple hypothesis correction, and no independent replication of these findings has occurred to date.
The fifth GWAS for DR was in Chinese subjects, and compared 570 controls defined as patients with T2D for more than 8 years without DR vs. 437 patients with T2D and PDR [48]. Severity of DR was classified as none, NPDR, and PDR based on the International Clinical Diabetic Retinopathy Disease Severity Scale. The cases and controls were similar with regards to duration of diabetes and HbA1c, the two strongest environmental influences on DR, but the study did not reveal any genome-wide significant loci. The sixth GWAS was in a Japanese population and used a three-step GWAS design to analyze a total of 837 T2D patients with DR and 1,149 without DR [49]. The severity of DR was based on fundus examination and graded as none, NPDR, or PDR based on an international clinical diabetic retinopathy/macular edema disease severity scale. The SNP with the smallest p-value from this study was rs9362054, which is located in an intron of RP1-90L14.1, but it was short of achieving genome-wide significance (p = 1.4 × 10-7).
The most recent GWAS was conducted in samples collected from non-Hispanic whites with T2D [50]. The discovery analysis was performed in 336 cases with sight-threatening DR and 508 controls with diabetes but no DR. Retinopathy status was determined from direct ophthalmic examination by the treating ophthalmologist, and was graded according to modified ETDRS criteria. The top-ranked SNP from the discovery cohort was rs3805931, with p = 2.66 x 10-7, but it was not associated with DR in the replication cohort. Only rs9896052 (p = 6.51 x 10-5 in the discovery cohort) showed a similar association in both T2D (p = 0.035) and T1D (p = 0.041), and was also associated in a third replication cohort from India (p = 0.016). The study-wide meta-analysis for this SNP reached genome-wide significance (p = 4.15 x 10-8). The GRB2 gene is located downstream of this variant, and a mouse model of retinopathy showed increased GRB2 expression in the retina.
Three studies have examined top signals from GWAS on DR in independent cohorts. In 2014, McAuley et al. genotyped 24 SNPs previously implicated in DR in 163 cases with T2D and severe NPDR or PDR and 300 controls with T2D for at least 5 years but no or mild DR [51]. Trained graders masked to clinical measures assessed images for the presence of retinopathy using the modified Airlie House classification as used in the ETDRS. In patients with severe NPDR/PDR and T1D or T2D, the authors reported rs1073203 in a dominant model (p = 0.005) and rs4838605 in an additive model (p = 0.047) to be associated with DR. The rs1073203 locus was initially identified in a T1D cohort [51].The association for rs4838605 with DR was identified previously in a Taiwanese DR study [46]. The allelic frequencies of this locus differ greatly by ethnicity [46], confirming that this association was replicable in a predominantly non-Hispanic white population, which suggested that this locus could be a trans-ethnic genetic marker for DR [51].
Hosseini et al. investigated the signals from large candidate gene studies and GWAS for both DR and diabetic kidney disease (DKD) in their own retinopathy samples [32]. Among 34 previous signals for DR and 55 previous loci for DKD, no association with severe DR was found after controlling for multiple testing. Of particular interest, rs1617640 in EPO was not significantly associated with DR status, combined severe DR-DKD phenotype, or time to severe DR. In 2015, Peng et al. genotyped 40 top signals from three GWAS on DR in Chinese patients with T2D, including 819 patients with DR and 1,153 patients without DR [52]. No SNPs were significantly associated with DR after adjustment for other DR risk factors and for multiple-hypothesis correction. In a subanalysis that compared patients with mild NPDR vs. those with severe NPDR or PDR, rs899036 was associated with severe DR (p = 0.009), but the results were short of attaining genome-wide significance after a meta-analysis with previous GWAS data (OR: 0.33; p = 5.84 × 10-7).
3. Diabetic kidney disease
3.1 Heritability and linkage studies
Familial aggregation of DKD has been shown in several ethnicities [10, 53-62]. In T1D, risk of DKD increased from 17-22% for patient whose siblings did not have DKD to 72-83% if a sibling had a DKD [10]. Familial aggregation appears to be particularly marked in African Americans [54] and Pima Indians [59, 60] compared to other ethnicities. In 1998, Imperatore and colleagues conducted a sibling-pair linkage analysis for DKD in Pima Indians with T2D, and found evidence of linkage at chromosome 7 with a LOD score of 2.7 [19]. In 2002, Vardali and colleagues found strong linkage at chromosome 18 with a LOD score 6.6 for DKD in a Turkish population [63]. In a genome scan of DKD in African Americans conducted in 2004, Bowden and colleagues revealed suggestive evidence for susceptibility loci on chromosomes 3q, 7p, and 18q with LOD scores 4.55, 3.59, and 3.72, respectively [64]. The Family Investigation of Nephropathy and Diabetes (FIND) in 2005 reinforced the evidence for DKD genes on chromosomes 3q, 7q, and 18q, and uncovered some evidence for a locus on chromosome 10p [65]. In 2007, FIND further reported that the strongest evidence of linkage to the DKD trait was specifically on chromosomes 7q21.3, 10p15.3, 14q23.1, and 18q22.3.
For albuminuria, the strongest linkage signals were on chromosomes 2q14.1, 7q21.1, and 15q26.3. In 2011, an analysis with the largest number of FIND samples available (3,972) reported linkage for DKD by ethnicity [66]. A chromosome 6 locus had a LOD score 3.09 in European Americans, and there was suggestive evidence for linkage to chromosome 7p in African American families. Regions on chromosomes 3p in African Americans, 7q in European Americans, 16q in African Americans, and 22q in Mexican Americans showed suggestive evidence of linkage for albuminuria. The linkage peak on chromosome 22q overlapped the MYH9/APOL1 gene region, which was previously implicated in African American diabetic and non-diabetic nephropathies [67, 68]. Overall, regions of linkage to DKD on chromosomes 7q, 10p, and 18q have been most consistent, making it important that genes underlying these peaks be particularly evaluated for their contribution to DKD susceptibility [69].
3.2 Candidate gene association studies
Genes that have a putative role in DKD have been investigated by several groups. These genes include EPO, protein kinase C (PKC), genes in the renin-angiotensin system, RAGE genes, and endothelial nitric oxide synthase (NOS3) [70]. We will highlight candidate gene investigations that were statistically most highly powered to detect associations.
EPO has been associated with both DR and DKD in three European-American cohorts [30]. The study showed that the T allele of rs1617640 in the EPO promoter was significantly associated with PDR. The allele was also found to be significantly associated with end-stage renal disease (ESRD) in the three cohorts (Utah: p = 1.91 ×10-3, GoKinD: p = 2.66 ×10-8, and Boston: p = 2.1×10-2) [30]. In 2012, the Genetics of Nephropathy, an International Effort (GENIE) consortium examined previously reported genetic associations with DKD in T1D [71]. GENIE consisted of 6,366 participants of European ancestry with T1D, either with or without DKD. The association of the EPO promoter polymorphism, rs1617640, with DKD was evaluated by de novo genotyping of the SNP. No significant associations were observed in the United Kingdom-Republic of Ireland (UK-ROI) or Finnish Diabetic Nephropathy (FinnDiane) collections of participants from GENIE, although the directions of effect were consistent with the original report. Fixed-effects meta-analysis of the association of rs1617640 with ESRD, including the previously reported cohorts (a total of 3,162 case and 3,845 control subjects across five separate cohorts of European and European-American ancestry), retained genome-wide statistical significance (p = 2 × 10-9).
PKC is an important regulating molecule in vascular function; its activation was suggested to induce the growth factors VEGF and TGF-β. [1] Human munc13 is a member of the PKC superfamily; it lacks the kinase domain and has a potential role in mediating some of the acute and chronic changes in mesangial cells caused by exposure to hyperglycemia [72]. In 2008, the European Rational Approach for the Genetics of Diabetic Complications (EURAGEDIC) study investigated 127 candidate genes for nephropathy. Only one SNP, rs2281999, which is located in the UNC13B gene, was significantly associated with nephropathy after correction for multiple testing (p = 1.79 × 10-5) [73]. In a cohort study from 2010, Ma et al. genotyped 18 common tag SNPs that span the PRKCB1 gene in 2,221 Chinese patients without renal disease at baseline [74]. Genetic variants in the PRKCB1 gene were independently associated with the development of ESRD in Chinese patients with T2D. The closely linked T allele of rs3760106 and G allele of rs2575390 (r2 = 0.98) showed the strongest association with ESRD, with hazard risk ratios of 2.25 (p = 0.03) and 2.26 (p = 0.003), respectively.
Wang et al. performed a meta-analysis of 14,108 DKD cases and 12,472 controls from 63 published studies [75]. When the authors included all ethnicities and both diabetes types, there was a significant association between an ACE insertion/deletion polymorphism and the risk of DKD. However, when the data were stratified by ethnicity and diabetes type, only Asians with T2D showed a significant association between the polymorphism and DKD.
The -374 T/A polymorphism in the gene encoding RAGE (AGER) was initially investigated in 3,334 T1D and T2D patients and 205 non-diabetic control subjects of Scandinavian origin [38]. The polymorphism was associated with DKD in both T1D and T2D, and in an HbA1c-dependent manner in the latter group. A meta-analysis to assess associations of other RAGE polymorphisms with DKD did not find any significant association between three RAGE gene polymorphisms (Gly82Ser, 1704G/T, 429T/C) and DKD risk. More recently, a meta-analysis of two polymorphisms in RAGE, -374T/A and -429T/C, and DKD risk was conducted [76]. Eight studies with 1,725 cases and 1,857 controls were included in the -374T/A polymorphism analysis. The main analysis indicated no association. Subgroup analyses in Caucasians and T2D patients also showed no association between the -374T/A polymorphism and DKD. When the -429C allele was examined using a recessive model, there was only a marginal association overall.
Nitric oxide (NO), a vasodilator molecule, is produced through the oxidation of L-arginine by endothelial nitric oxide synthase (eNOS) [77]. In 2003, Nagase et al. showed that some intronic polymorphisms in eNOS may change its transcriptional activity. They also found that some exonic polymorphisms may alter the three-dimensional structure of the enzyme and affect the progression of renal disease via decreased NO synthesis [78]. In a meta-analysis of 18 eNOS studies, the eNOS-4b/a polymorphism was significantly associated with an overall increased risk of DKD across all models examined [79]. Subgroup analysis revealed a significant association between the eNOS-4b/a polymorphism and DKD in Asian populations, especially in the Chinese population, but not in non-Asians.
In 2011, Mooyaart et al. performed a meta-analysis of 671 genetic association studies investigating DKD to assess the pooled effect of each genetic variant reproducibly associated with DKD [80]. They identified 34 replicated genetic variants. Of these, 21 remained significantly associated with DKD in a random-effects meta-analysis. These variants were in or near the following genes: ACE, AKR1B1 (two variants), APOC1, APOE, EPO, NOS3 (two variants), HSPG2, VEGFA, FRMD3 (two variants), CARS (two variants), UNC13B, CPVL and CHN2, and GREM1, plus four variants that were not near genes. Three additional variants were not significantly associated with DKD in the whole population after meta-analyses, but were associated in one subgroup: ELMO1 (Asians), CCR5 (Asians), and CNDP1 (T2D).
3.3 Genome wide association studies
Genome-wide association approaches have been attempted for DKD, and are starting to reveal some loci that can be independently replicated (for a summary of study results see Table 2). Some of the initial studies were relatively small, and did not sample the genome very densely. In 2007, Maeda et al. performed a low-coverage GWAS (80,000 SNPs genotyped), comparing 94 nephropathy cases and 94 controls [61]. They identified SLC12A3 and engulfment and cell motility 1 gene (ELMO1) as candidate genes for DKD. A larger, but lower condensed (6,000 microsatellites) GWAS in a cohort of Irish patients with T1D (200 cases and 200 controls) revealed nominally significant associations with two markers on chromosome 10 (D10S558, p = 0.005; D10S1435, p = 0.016). A 100,000 SNP GWAS in Pima Indians conducted on pooled genomic DNA from 105 cases and 102 controls identified a potential association with PVT1 [81].
Table
2.
Top signals from genome-wide associations studies examining diabetic kidney disease |
|
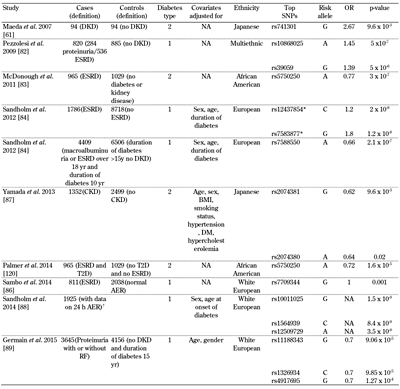 |
|
Legend:
* After combined meta-analysis (meta-analysis results for GENIE discovery cohorts + meta-analysis results for independent replication cohorts), these signals reached genome-wide significance in the analysis. † This study selected patients who passed the genetic quality control thresholds, and had 24-h albumin exertion rate (AER). Abbreviations: DKD – diabetic kidney disease, OR – odd ratio, NA – not available, ESRD – end-stage renal disease, CKD – chronic kidney disease, T2D – type 2 diabetes, AER – albumin exertion rate. |
|
In 2009, Pezzolesi et al. genotyped approximately 360,000 SNPs in 820 case subjects (284 with proteinuria and 536 with ESRD) and 885 control subjects (T1D for at least 15 years with normoalbuminuria) [82]. A total of 13 SNPs located in four genomic loci were associated with diabetic nephropathy (p < 1 x 10-5). The strongest association was in FRMD3 (p = 5.0 x 10-7). Another top association was at the cysteinyl-tRNA synthetase 9 (CARS) locus (p = 3.1 x 10-6). The authors demonstrated expression of both FRMD3 and CARS in the human kidney.
A GWAS aimed at identifying genes associated with DKD in African Americans was performed by adjustment for admixture [83]. In a sub-analysis, cases with ≥100 mg/dl of proteinuria were compared to T2D subjects without nephropathy. It appeared that no SNP was genome-wide significant. The authors identified SNPs in RPS12 (rs9493454) and AUH (rs7735506) as being strongly associated in this DKD-specific subanalysis (p = 8.79×10-4 and p = 2.57×10-4, OR (95% CI) = 1.20 (1.08-1.34) and OR (95% CI) = 0.78 (0.69-0.89), respectively).
In 2012, the GENIE consortium performed a GWAS of T1D and DKD comprising approximately 2.4 million SNPs included in 6,691 individuals [84]. The primary phenotype of interest was diabetic nephropathy, defined by the presence of persistent macroalbuminuria or ESRD in individuals aged over 18 with T1D for at least 10 years. Controls were defined as individuals with T1D for at least 15 years, but without any clinical evidence of kidney disease. After additional genotyping of 41 top-ranked SNPs, representing 24 independent signals in 5,873 individuals, combined meta-analysis revealed association of two SNPs with ESRD: rs7583877 in AFF3 (p = 1.2 × 10-8 ) and an intergenic SNP on chromosome 15q26 between the genes RGMA and MCTP2, rs12437854 (p = 2.0 × 10-9). Functional data suggested that AFF3 influences renal tubule fibrosis via the transforming growth factor-beta 1 (TGF-β1) pathway. The strongest association with DKD as a primary phenotype was seen for an intronic SNP in the ERBB4 gene (rs7588550, p = 2.1 × 10-7), which is located in the same intron as a variant with cis-eQTL expression of ERBB4, and associated with differential expression of T2D and DKD [84]. FinnDiane and GENIE performed a subsequent GWAS to determine whether sex-specific genetic risk factors for ESRD exist [85]. The authors performed comparisons between T1D patients who had ESRD and T1D controls who had no evidence of DKD despite long duration of diabetes. ESRD was defined as the need for dialysis treatment or having received a kidney transplant, and the minimum duration of T1D was 10 years. In FinnDiane, a common variant, rs4972593 on chromosome 2q31.1, was associated with ESRD in women (p = 3.02 × 10-8), but not in men (p = 0.78). This association was replicated in three independent T1D cohorts with a combined p-value of 0.02, and remained significant for women when meta-analysis was performed between FinnDiane and the replications cohorts (p = 3.85 × 10-8).
Subsequently, the results were also reported for a GWAS in which 3,464 patients with T1D from the FinnDiane Study were the discovery cohort. Replication was performed with 4,263 T1D patients from Steno Diabetes Centre, UK-ROI, and GoKinD United States [86]. The samples in this analysis overlapped significantly with the previous FinnDiane/GENIE GWAS analyses, but employed a naïve Bayesian marker selection strategy that is complementary to that of conventional GWAS models. Diabetic nephropathy was defined as persistent macroalbuminuria (urinary albumin excretion rate (AER) ≥200 μg/min or ≥300 mg/24 h, or a urinary albumin/creatinine ratio ≥25 mg/mmol for men and ≥35 mg/mmol for women, or dipstick ≥1) in two out of three consecutive measurements and an absence of other known kidney or urinary tract diseases. Microalbuminuria was defined as 20 ≤ AER < 200 μg/min or 30 ≤ AER < 300 mg/24 h or 2.5 ≤ albumin-creatinine ratio (ACR) < 25 mg/mmol for men and 3.5 ≤ ACR < 35 mg/mmol for women in two out of three consecutive urine collections. Absence of nephropathy was defined as a persistently normal AER (AER <20 μg/min or <30 mg/24 h, or ACR <2.5 mg/mmol for men or ACR <3.5 mg/mmol for women), after at least 15 years of diabetes. ESRD was defined as ongoing dialysis treatment or a past kidney transplant. In the Steno, UK-ROI, and GoKinDUS studies, the enrolled patients with a normal AER were not treated with ACE inhibitors (or angiotensin-II receptor blockers in the Steno and UK-ROI studies). In the Steno study, the patients with nephropathy had concurrent retinopathy, with the exception of four cases, whose diagnosis of diabetic glomerulopathy was verified by kidney biopsy. No patients with microalbuminuria were included in the UK-ROI and GoKinD US studies. This strategy identified the following five genetic loci:
1. rs12137135 (p = 1.29 × 10-5)
2. rs17709344 (p = 2.44 × 10-5)
3. rs1670754 (p = 7.71 × 10-6)
4. rs12917114 (p = 2.1 × 10-7)
5. rs2838302 (p = 0.0002)
These loci were potentially associated with ESRD in the FinnDiane study. rs17709344 is located between the RGMA and MCTP2 genes, and tags the previously identified rs12437854. An association between ESRD and rs17709344 was consistent regarding direction of effect for all replication cohorts (p = 0.012). rs12917114 near SEMA6D was also associated with ESRD in the replication cohorts under the genotypic model (p = 0.049). rs12137135 upstream of WNT4 was associated with ESRD only in the Steno samples.
In 2013, Yamada et al. reported on a GWAS including 3,851 Japanese individuals from three independent subject panels [87]. Subject panels A, B, and C comprised 252, 910, and 190 individuals with chronic kidney disease (CKD) and 249, 838, and 1,412 controls, respectively. Approximately half of the patients had diabetes, either T1D or T2D. The GWAS for CKD was performed in subject panel A. The selection criteria for subject panel A were as follows:
1. Estimated glomerular filtration rate (eGFR) for subjects with CKD: <40 ml/min per 1.73 m2
2. eGFR for controls: ≥90 ml/min per 1.73 m2
3. Age of control subjects: ≥64 years
4. No renal disease for control subjects
5. No or only minor health problems for control subjects
SNPs within chromosome 3q28, ALPK1, FAM78B, and UMODL1 were significantly (false discovery rate <0.05) associated with CKD. The relation of SNPs at these five loci with CKD was examined in subject panel B; rs9846911 at 3q28 (p = 4.5 × 10-6) was significantly associated with CKD in all individuals, and rs2074381 (p = 0.004) and rs2074380 (p = 0.005) in ALPK1 were associated with CKD in individuals with diabetes mellitus. These three SNPs were further examined in subject panel C, revealing that rs2074381 (p = 0.02) and rs2074380 (p = 0.01) were significantly associated with CKD. For subject panels B and C in combination, rs9846911 (p = 0.0007) was significantly associated with CKD in all individuals and rs2074381 (p = 0.01) and rs2074380 (p = 0.02) were associated with CKD in diabetic individuals.
In 2014, Sandholm performed a GWAS including 1,925 patients with T1D to analyze the 24-hour AER as a continuous trait [88]. The analysis was stratified by the use of antihypertensive medication. The narrow-sense heritability, captured with their genotyping platform, was estimated to explain 27.3% of the total AER variability and 37.6% after adjustment for covariates. In the discovery stage, SNPs in the GLRA3 gene were strongly associated with albuminuria, with the strongest association found for rs10011025 (p = 1.5 x 10-9), but this locus could not be replicated in independent cohorts. In 2015, Germain et al. performed a GWAS comparing T1D cases with proteinuria (with or without renal failure) with control patients who have had diabetes for more than 15 years and no evidence of renal disease [89]. No SNPs tested in a discovery cohort consisting of 683 cases and 779 controls reached genome-wide statistical significance.
3.4 Exome sequencing
In 2014, exome sequencing of coding variants in genes with prior evidence for association with ESRD or nephropathy was performed; the study included 5,045 African Americans (3,324 cases with T2D-associated ESRD and non-T2D-associated ESRD, and 1,721 diabetic and non-diabetic controls) and 1,465 European Americans (568 T2D-ESRD cases and 897 controls) [90]. In African Americans, several SNPs were nominally associated with T2D-ESRD and all-cause ESRD with p-values ranging from 1.8 × 10-4 to 0.04. Haplotype analysis of common and coding variants increased evidence of association at the OR2L13 and APOL1 loci (p = 6.2 × 10-5 and 4.6 × 10-5, respectively). SNPs replicated in European Americans were found in OR2AK2, LIMK2, and APOL2 with p-values ranging from 0.001 to 0.04.
While several promising loci have emerged from GWAS in DKD, further validation of these results is required to definitively establish these loci as risk factors for nephropathy. There are other ongoing initiatives in larger samples sizes, including in the Juvenile Diabetes Research Foundation-sponsored Diabetic Nephropathy Collaborative Research Initiative (DNCRI), FIND, and Surrogate Markers for Micro- and Macrovascular Hard Endpoints for Innovative Diabetes Tools (SUMMIT). The results of these studies should help to further clarify the genetic underpinnings of DKD.
4. Diabetic neuropathy
The prevalence of diabetic neuropathy varies from 14% to 63%, depending upon the type of population and the criteria used to define the disease [91]. There have been fewer genetic studies for diabetic neuropathy as compared to DR and DKD. Diabetic neuropathy encompasses a broad spectrum of clinical entities. Consequently, the selection of a well-defined phenotype for multicenter studies has been a challenge. To date, two major forms of the disease have been examined: peripheral neuropathy and cardiovascular autonomic neuropathy.
4.1 Linkage studies
A linkage study on neuropathy was focused on chromosome 12q24 based on previous reports of a gene at this locus being associated with DR, DKD, and T2D [92-94]. 52 Italian families were examined using both nonparametric and parametric linkage analyses for diabetic neuropathy [95]. Evidence of linkage of chromosome 12q24 locus with diabetic neuropathy was found. This locus that includes proteasome modulator 9 (PSMD9), a transcriptional regulator of the insulin gene, had a LOD score of 3.34 (p = 0.0009).
4.2 Candidate gene association studies
Some of the candidate genes that have been studied in the context of DR and DKD have also been examined in diabetic neuropathy. Zhang et al. evaluated the relationship between a 936C/T mutation at the 3'-untranslated region of the human VEGF gene and diabetic peripheral neuropathy [96]. They evaluated 204 cases with diabetic peripheral neuropathy, 184 cases with T2D without neuropathy and 240 healthy controls. The T allele frequency in the diabetic peripheral neuropathy group was lower than in T2D control (p = 0.001). In 154 Italian patients with T2D and 171 healthy controls, an association between the TCF7L2 rs7903146 variant and the presence of cardiovascular autonomic neuropathy has been reported (p = 0.02) [34]. Samples from subjects with diabetic neuropathy have also been evaluated for mutations in the AKR1B1 gene (exon 1) [97]. Of 50 diabetic neuropathy patient samples analyzed, 10 revealed mutations. Wu et al. performed a meta-analysis comprising a total of seven case-control studies, including 1,316 cases and 1,617 controls, to evaluate the effects of ACE I/D polymorphisms in the development of diabetic peripheral neuropathy [98]. They found that the ACE I/D polymorphism was associated with increased risk of diabetic peripheral neuropathy (p = 0.006) [99]. Tang et al. examined the association between the T allele of rs1050450 in glutathione peroxidase-1 (GPx-1) and peripheral neuropathy in two cross-sectional samples of subjects with diabetes: 1) 773 Caucasian subjects were genotyped by the University College London Diabetes and Cardiovascular disease Study (UDACS) and 2) 382 Caucasian subjects from the Ealing Diabetes Study (EDS) [100]. The ORs for peripheral neuropathy in T allele carriers compared to the CC genotype were 1.61 (95% CI: 1.10-2.28, p = 0.01) in UDACS and 1.95 (95% CI: 1.11-3.42, p = 0.02) in EDS. This T allele is associated with reduced enzyme activity.
Variations in microRNAs (miRNAs) have also been examined in neuropathy. In 2014, Ciccacci et al. evaluated a possible involvement of genetic polymorphisms in miRNA regions in the susceptibility to diabetic peripheral neuropathy and cardiovascular autonomic neuropathy [101]. Nine polymorphisms in miRNA (MIR375, MIR27a, MIR130b, MIR124a (2 SNPs), MIR128a, MIR194a, and MIR146a) genes were studied in a sample of 132 T2D patients analyzed for diabetic peripheral neuropathy and 128 T2D patients analyzed for cardiovascular autonomic neuropathy. Associations of MIR128a (p = 0.007) and MIR146a SNPs (p = 0.032) with peripheral neuropathy susceptibility and of MIR146a (p = 0.041) and MIR27a (p = 0.023) with cardiovascular autonomic susceptibility were found, but correction for multiple hypothesis testing was not performed.
4.3 Genome-wide association studies
There has only been one GWAS focusing on diabetic neuropathy to date. The Genetics of Diabetes Audit and Research Tayside (GoDARTS) group performed a GWAS for neuropathic pain [102]. 6,927 diabetic patients were included, and cases of neuropathic pain were defined as patients with a prescription history of at least one of five drugs specifically indicated for the treatment of neuropathic pain, and in whom the monofilament test result was positive for sensory neuropathy in at least one foot. Controls were diabetic individuals who did not have a record of receiving any opioid analgesics. They identified a cluster of associations on chromosome 8p21.3, next to glial cell line-derived neurotropic factor family receptor alpha 2 (GFRA2), with a lowest p-value of 1.77 × 10-7 at rs17428041.
5. Epigenetic factors
Epigenetic factors have been examined primarily in DKD [103-105]. Epigenetic mechanisms such as post-translational modifications (PTM) in chromatin and histones as well as DNA methylation (DNAme) may have a role in diabetic microvascular complications. Hyperglycemia, growth factors, oxidant stress, and inflammatory factors in diabetes can cause changes in these epigenetic mechanisms, and alter the expression of pathological genes in target cells [106].
FinnDiane found an association between an exonic SNP, rs17353856, in the SUV39H1 histone methyltransferase gene with DR, and a trend toward an association with DKD [107]. One study assessed DNAme in whole blood genomic DNA obtained from patients with DKD in relation to those without DKD. There was differential methylation in a number of genes, including UNC13B which has been previously linked with DKD. Pirola et al. reported that hyperglycemia led to changes in DNAme at key genes involved in endothelial cell dysfunction [108]. Kim et al. found that control of hyperglycemia induced alterations in DNAme at key genes involved in dysfunction of Schwann cells [109]. An investigation of methylome-wide loci for association with DKD in individuals with T1D found differences in methylation between 150 cases with DKD and 100 diabetic controls without renal disease [110]. Following validation and replication, CCNL1 and ZNF187 had differentially regulated loci (p < 10-8), with evidence also established for AFF3. Finally, Swan et al. in 2015 studied methylation profiles in genes related to mitochondrial function to assess whether differences in these epigenetic features were associated with DKD in people with T1D [111]. A case-control association study was performed in 196 individuals with DKD and 246 individuals without renal disease. They reported that fifty-four cytosine-phosphate-guanine probes across 51 unique genes were significantly associated with DKD (p ≤ 10-8) across both the 450K and the 27K methylation arrays. A subanalysis, employing the 450K array, identified 755 cytosine-phosphate-guanine probes in 374 genes that were significantly associated with ESRD (p ≤ 10-8). Forty-six of the top-ranked variants for DKD were also identified as being differentially methylated in individuals with ESRD. The largest change in methylation (Δβ = 0.2) was observed for cg03169527 in the TAMM41 gene on chromosome 3p25.2. Three genes, PMPCB, TSFM, and AUH, had differential methylation at multiple cytosine-phosphate-guanine sites each (p < 10-12).
There was early evidence that miRNAs may impact diabetic microvascular complications through epigenetic mechanisms [112]. Changes in miRNA expression have been showed in diabetic eyes; miRNAs in the eye, including miR200b (one of the VEGF-regulating miRNAs) is downregulated [113]. Upregulation of miR-29b in the early stages of diabetes is considered to be protective against apoptosis of the retinal ganglion cells [114]. miRNAs also play a role in TGF-β signaling related to the pathogenesis of DKD; key miRNAs such as miR-192, miR-216a, miR-217, and miR-377 appeared to be upregulated, resulting in increased fibronectin and collagen expression [115, 116].
6. Conclusions
Genetic discovery in microvascular complications of diabetes is more advanced for DKD and DR than for diabetic neuropathy. Relatively little has been done to study the genetics of neuropathy. Thus far there has been a lack of reproducible associations for DKD and DR. This can be explained in large part by the relatively limited samples that have been evaluated to date. However, there are also other potential explanations for this situation.
Firstly, only common variation has been comprehensively examined for microvascular complications thus far. The role of rare variants remains to be explored, and it may be that these complex phenotypes are due to the collective effect of multiple rare variants with moderate to high penetrance [117]. In particular, association tests that group rare and common variants by gene or functional unit may help to uncover some of the variation that cannot be found by just examining common variants.
Variability in the microvascular phenotype definitions is another weakness in this field. Some of the case definitions used in DR and DKD GWAS include milder degrees of retinopathy and nephropathy that bias the results towards the null. Cases that are defined as having more advanced levels of complications are likely to avoid this bias. There are also important sources of heterogeneity that might contribute to inconsistencies in findings between the studies. For example DR ascertainment was performed differently, with one GWAS relying on questionnaire documentation of eye laser treatment [15, 44, 45], which has been validated as a measure of severe retinopathy, while other GWAS used fundus examination and/or photography [43, 48, 49, 118]. Also, the various GWAS for DR and DKD have examined different case definitions including PDR, NPDR, DME, proteinuria, AER, and ESRD. In some studies, participants with NPDR and microproteinuria were classified as controls [43-45, 87], whereas in other studies they were considered cases [46, 61, 82, 89].
Finally, the studies have not consistently defined controls strictly with regards to duration of diabetes [49, 61, 87]. Duration of diabetes has a strong impact on development of complications, as does glycemic control, and these factors need to be taken into account. Inclusion of participants with short durations of diabetes can cause misclassification of controls, and thus bias results to the null. Ideally, controls would be participants with very long durations of diabetes and no DR or DKD. Based on the recognition that these individuals are a small subset of the diabetic population, controls have often been defined more loosely than this to achieve a critical control sample size in today’s studies [119].
The key to future success in studies of diabetic microvascular complications will be in collaborative efforts to bring together large sample sizes with well-defined case and control definitions that account for duration of diabetes and glycemic control, and examine both common and rare variation comprehensively. Such collaborative efforts are already underway, and hold the promise for improving our understanding of the genetic factors that influence the susceptibility of diabetic patients to developing microvascular complications.
Disclosures: The authors report no conflict of interests.
References
- Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA 2002. 288(20):2579-2588. [DOD] [CrossRef]
- Pirart J. Diabetes mellitus and its degenerative complications: a prospective study of 4,400 patients observed between 1947 and 1973 (3rd and last part). Diabete Metab 1977. 3(4):245-256. [DOD]
- Stolar M. Glycemic control and complications in type 2 diabetes mellitus. Am J Med 2010. 123(3 Suppl):S3-S11. [DOD] [CrossRef]
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998. 317(7160):703-713. [DOD] [CrossRef]
- Clustering of long-term complications in families with diabetes in the diabetes control and complications trial. The Diabetes Control and Complications Trial Research Group. Diabetes 1997. 46(11):1829-1839. [DOD] [CrossRef]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001. 414(6865):813-820. [DOD] [CrossRef]
- Heilig CW, Concepcion LA, Riser BL, Freytag SO, Zhu M, Cortes P. Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J Clin invest 1995. 96(4):1802-1814. [DOD] [CrossRef]
- Lachin JM, Genuth S, Nathan DM, Zinman B, Rutledge BN. Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial - revisited. Diabetes 2008. 57(4):995-1001. [DOD] [CrossRef]
- Hirsch IB, Brownlee M. Beyond hemoglobin A1c - need for additional markers of risk for diabetic microvascular complications. JAMA 2010. 303(22):2291-2292. [DOD] [CrossRef]
- Quinn M, Angelico MC, Warram JH, Krolewski AS. Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia 1996. 39(8):940-945. [DOD] [CrossRef]
- Leslie RD, Pyke DA. Diabetic retinopathy in identical twins. Diabetes 1982. 31(1):19-21. [DOD] [CrossRef]
- Pyke DA, Tattersall RB. Diabetic retinopathy in identical twins. Diabetes 1973. 22(8):613-618. [DOD] [CrossRef]
- Hietala K, Forsblom C, Summanen P, Groop PH. Heritability of proliferative diabetic retinopathy. Diabetes 2008. 57(8):2176-2180. [DOD] [CrossRef]
- Looker HC, Nelson RG, Chew E, Klein R, Klein BE, Knowler WC, Hanson RL. Genome-wide linkage analyses to identify Loci for diabetic retinopathy. Diabetes 2007. 56(4):1160-1166. [DOD] [CrossRef]
- Rema M, Saravanan G, Deepa R, Mohan V. Familial clustering of diabetic retinopathy in South Indian Type 2 diabetic patients. Diabet Med 2002. 19(11):910-916. [DOD] [CrossRef]
- Zhang X, Gao Y, Zhou Z, Wang J, Zhou Q, Li Q. Familial clustering of diabetic retinopathy in Chongqing, China, type 2 diabetic patients. Europ J Ophthalmol 2010. 20(5):911-918. [DOD]
- Hallman DM, Huber JC Jr, Gonzalez VH, Klein BE, Klein R, Hanis CL. Familial aggregation of severity of diabetic retinopathy in Mexican Americans from Starr County, Texas. Diabetes Care 2005. 28(5):1163-1168. [DOD] [CrossRef]
- Arar NH, Freedman BI, Adler SG, Iyengar SK, Chew EY, Davis MD, Satko SG, Bowden DW, Duggirala R, Elston RC, et al. Heritability of the severity of diabetic retinopathy: the FIND-Eye study. Invest Ophthalmol Vis Sci 2008. 49(9):3839-3845. [DOD] [CrossRef]
- Imperatore G, Hanson RL, Pettitt DJ, Kobes S, Bennett PH, Knowler WC. Sib-pair linkage analysis for susceptibility genes for microvascular complications among Pima Indians with type 2 diabetes. Pima Diabetes Genes Group. Diabetes 1998. 47(5):821-830. [DOD] [CrossRef]
- Hallman DM, Boerwinkle E, Gonzalez VH, Klein BE, Klein R, Hanis CL. A genome-wide linkage scan for diabetic retinopathy susceptibility genes in Mexican Americans with type 2 diabetes from Starr County, Texas. Diabetes 2007. 56(4):1167-1173. [DOD] [CrossRef]
- Kuo JZ, Wong TY, Rotter JI. Challenges in elucidating the genetics of diabetic retinopathy. JAMA Ophthalmol 2014. 132(1):96-107. [DOD] [CrossRef]
- Abhary S, Hewitt AW, Burdon KP, Craig JE. A systematic meta-analysis of genetic association studies for diabetic retinopathy. Diabetes 2009. 58(9):2137-2147. [DOD] [CrossRef]
- Liew G, Klein R, Wong TY. The role of genetics in susceptibility to diabetic retinopathy. Int Ophthalmol Clin 2009. 49(2):35-52. [DOD] [CrossRef]
- Osaadon P, Fagan XJ, Lifshitz T, Levy J. A review of anti-VEGF agents for proliferative diabetic retinopathy. Eye 2014. 28(5):510-520. [DOD] [CrossRef]
- Awata T, Kurihara S, Takata N, Neda T, Iizuka H, Ohkubo T, Osaki M, Watanabe M, Nakashima Y, Inukai K, et al. Functional VEGF C-634G polymorphism is associated with development of diabetic macular edema and correlated with macular retinal thickness in type 2 diabetes. Biochem Biophys Res Commun 2005. 333(3):679-685. [DOD] [CrossRef]
- Al-Kateb H, Mirea L, Xie X, Sun L, Liu M, Chen H, Bull SB, Boright AP, Paterson AD. Multiple variants in vascular endothelial growth factor (VEGFA) are risk factors for time to severe retinopathy in type 1 diabetes: the DCCT/EDIC genetics study. Diabetes 2007. 56(8):2161-2168. [DOD] [CrossRef]
- Bonnefond A, Saulnier PJ, Stathopoulou MG, Grarup N, Ndiaye NC, Roussel R, Nezhad MA, Dechaume A, Lantieri O, Hercberg S, et al. What is the contribution of two genetic variants regulating VEGF levels to type 2 diabetes risk and to microvascular complications? Plos One 2013. 8(2):e55921. [DOD]
- Han L, Zhang L, Xing W, Zhuo R, Lin X, Hao Y, Wu Q, Zhao J. The associations between VEGF gene polymorphisms and diabetic retinopathy susceptibility: a meta-analysis of 11 case-control studies. J Diabetes Res 2014. 2014:805801. [DOD]
- Kaidonis G, Burdon KP, Gillies MC, Abhary S, Essex RW, Chang JH, Pal B, Pefkianaki M, Daniell M, Lake S, et al. Common sequence variation in the VEGFC gene Is associated with diabetic retinopathy and diabetic macular edema. Ophthalmology 2015. In press. [DOD]
- Tong Z, Yang Z, Patel S, Chen H, Gibbs D, Yang X, Hau VS, Kaminoh Y, Harmon J, Pearson E, et al. Promoter polymorphism of the erythropoietin gene in severe diabetic eye and kidney complications. Proc Nat Acad Sci USA 2008. 105(19):6998-7003. [DOD] [CrossRef]
- Abhary S, Burdon KP, Casson RJ, Goggin M, Petrovsky NP, Craig JE. Association between erythropoietin gene polymorphisms and diabetic retinopathy. Arch Ophthalmol 2010. 128(1):102-106. [DOD] [CrossRef]
- Hosseini SM, Boright AP, Sun L, Canty AJ, Bull SB, Klein BE, Klein R, Paterson AD. The association of previously reported polymorphisms for microvascular complications in a meta-analysis of diabetic retinopathy. Hum Genet 2015. 134(2):247-257. [DOD] [CrossRef]
- Luo J, Zhao L, Chen AY, Zhang X, Zhu J, Zhao J, Ouyang H, Luo H, Song Y, Lee J, et al. TCF7L2 variation and proliferative diabetic retinopathy. Diabetes 2013. 62(7):2613-2617. [DOD] [CrossRef]
- Ciccacci C, Di Fusco D, Cacciotti L, Morganti R, D'Amato C, Novelli G, Sangiuolo F, Spallone V, Borgiani P. TCF7L2 gene polymorphisms and type 2 diabetes: association with diabetic retinopathy and cardiovascular autonomic neuropathy. Acta Diabetol 2013. 50(5):789-799. [DOD] [CrossRef]
- Buraczynska M, Swatowski A, Markowska-Gosik D, Kuczmaszewska A, Ksiazek A. Transcription factor 7-like 2 (TCF7L2) gene polymorphism and complication/comorbidity profile in type 2 diabetes patients. Diabetes Res Clin Pract 2011. 93(3):390-395. [DOD] [CrossRef]
- Schmidt AM, Hofmann M, Taguchi A, Yan SD, Stern DM. RAGE: a multiligand receptor contributing to the cellular response in diabetic vasculopathy and inflammation. Semin Thromb Hemost 2000. 26(5):485-493. [DOD] [CrossRef]
- Ramprasad S, Radha V, Mathias RA, Majumder PP, Rao MR, Rema M. Rage gene promoter polymorphisms and diabetic retinopathy in a clinic-based population from South India. Eye 2007. 21(3):395-401. [DOD] [CrossRef]
- Lindholm E, Bakhtadze E, Sjogren M, Cilio CM, Agardh E, Groop L, Agardh CD. The -374 T/A polymorphism in the gene encoding RAGE is associated with diabetic nephropathy and retinopathy in type 1 diabetic patients. Diabetologia 2006. 49(11):2745-2755. [DOD] [CrossRef]
- Kang P, Tian C, Jia C. Association of RAGE gene polymorphisms with type 2 diabetes mellitus, diabetic retinopathy and diabetic nephropathy. Gene 2012. 500(1):1-9. [DOD] [CrossRef]
- Niu W, Qi Y, Wu Z, Liu Y, Zhu D, Jin W. A meta-analysis of receptor for advanced glycation end products gene: four well-evaluated polymorphisms with diabetes mellitus. Mol Cell Endocrinol 2012. 358(1):9-17. [DOD] [CrossRef]
- Sobrin L, Green T, Sim X, Jensen RA, Tai ES, Tay WT, Wang JJ, Mitchell P, Sandholm N, Liu Y, et al. Candidate gene association study for diabetic retinopathy in persons with type 2 diabetes: the Candidate gene Association Resource (CARe). Invest Ophthalmol Vis Sci 2011. 52(10):7593-7602. [DOD] [CrossRef]
- Penman A, Hoadley S, Wilson JG, Taylor HA, Chen CJ, Sobrin L. P-selectin plasma levels and genetic variant associated with diabetic retinopathy in African Americans. Am J Ophthalmol 2015. 159(6):1152-1160. [DOD] [CrossRef]
- Fu YP, Hallman DM, Gonzalez VH, Klein BE, Klein R, Hayes MG, Cox NJ, Bell GI, Hanis CL. Identification of diabetic retinopathy genes through a genome-wide association study among Mexican-Americans from Starr County, Texas. J Ophthalmol 2010. 2010. [DOD]
- Grassi MA, Tikhomirov A, Ramalingam S, Below JE, Cox NJ, Nicolae DL. Genome-wide meta-analysis for severe diabetic retinopathy. Hum Mol Genet 2011. 20(12):2472-2481. [DOD] [CrossRef]
- Grassi MA, Tikhomirov A, Ramalingam S, Lee KE, Hosseini SM, Klein BE, Klein R, Lussier YA, Cox NJ, Nicolae DL. Replication analysis for severe diabetic retinopathy. Invest Ophthalmol Vis Sci 2012. 53(4):2377-2381. [DOD] [CrossRef]
- Huang YC, Lin JM, Lin HJ, Chen CC, Chen SY, Tsai CH, Tsai FJ. Genome-wide association study of diabetic retinopathy in a Taiwanese population. Ophthalmology 2011. 118(4):642-648. [DOD] [CrossRef]
- Lin HJ, Huang YC, Lin JM, Wu JY, Chen LA, Tsai FJ. Association of genes on chromosome 6, GRIK2 , TMEM217 and TMEM63B (linked to MRPL14 ) with diabetic retinopathy. Ophthalmologica 2013. 229(1):54-60. [DOD] [CrossRef]
- Sheu WH, Kuo JZ, Lee IT, Hung YJ, Lee WJ, Tsai HY, Wang JS, Goodarzi MO, Klein R, Klein BE, et al. Genome-wide association study in a Chinese population with diabetic retinopathy. Hum Mol Genet 2013. 22(15):3165-3173. [DOD] [CrossRef]
- Awata T, Yamashita H, Kurihara S, Morita-Ohkubo T, Miyashita Y, Katayama S, Mori K, Yoneya S, Kohda M, Okazaki Y, et al. A genome-wide association study for diabetic retinopathy in a Japanese population: potential association with a long intergenic non-coding RNA. Plos One 2014. 9(11):e111715. [DOD] [CrossRef]
- Burdon KP, Fogarty RD, Shen W, Abhary S, Kaidonis G, Appukuttan B, Hewitt AW, Sharma S, Daniell M, Essex RW, et al. Genome-wide association study for sight threatening diabetic retinopathy reveals association with genetic variation near the GRB2 gene. Diabetologia 2015. In press. [DOD]
- McAuley AK, Wang JJ, Dirani M, Connell PP, Lamoureux E, Hewitt AW. Replication of genetic loci implicated in diabetic retinopathy. Invest Ophthalmol Vis Sci 2014. 55(3):1666-1671. [DOD] [CrossRef]
- Peng D, Wang J, Zhang R, Jiang F, Tang S, Chen M, Yan J, Sun X, Wang S, Wang T, et al. Common variants in or near ZNRF1, COLEC12, SCYL1BP1 and API5 are associated with diabetic retinopathy in Chinese patients with type 2 diabetes. Diabetologia 2015. In press. [DOD]
- Seaquist ER, Goetz FC, Rich S, Barbosa J. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med 1989. 320(18):1161-1165. [DOD] [CrossRef]
- Freedman BI, Spray BJ, Tuttle AB, Buckalew VM Jr. The familial risk of end-stage renal disease in African Americans. Am J Kidney Dis 1993. 21(4):387-393. [DOD] [CrossRef]
- Spray BJ, Atassi NG, Tuttle AB, Freedman BI. Familial risk, age at onset, and cause of end-stage renal disease in white Americans. J Am Soc Nephrol 1995. 5(10):1806-1810. [DOD]
- O'Dea DF, Murphy SW, Hefferton D, Parfrey PS. Higher risk for renal failure in first-degree relatives of white patients with end-stage renal disease: a population-based study. Am J Kidney Dis 1998. 32(5):794-801. [DOD] [CrossRef]
- Forsblom CM, Kanninen T, Lehtovirta M, Saloranta C, Groop LC. Heritability of albumin excretion rate in families of patients with Type II diabetes. Diabetologia 1999. 42(11):1359-1366. [DOD] [CrossRef]
- Canani LH, Gerchman F, Gross JL. Familial clustering of diabetic nephropathy in Brazilian type 2 diabetic patients. Diabetes 1999. 48(4):909-913. [DOD] [CrossRef]
- Nelson RG, Newman JM, Knowler WC, Sievers ML, Kunzelman CL, Pettitt DJ, Moffett CD, Teutsch SM, Bennett PH. Incidence of end-stage renal disease in type 2 (non-insulin-dependent) diabetes mellitus in Pima Indians. Diabetologia 1988. 31(10):730-736. [DOD] [CrossRef]
- Pettitt DJ, Saad MF, Bennett PH, Nelson RG, Knowler WC. Familial predisposition to renal disease in two generations of Pima Indians with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1990. 33(7):438-443. [DOD] [CrossRef]
- Maeda S, Osawa N, Hayashi T, Tsukada S, Kobayashi M, Kikkawa R. Genetic variations associated with diabetic nephropathy and type II diabetes in a Japanese population. Kidney Int Suppl 2007. 2007(106):S43-S48. [DOD]
- Vijay V, Snehalatha C, Shina K, Lalitha S, Ramachandran A. Familial aggregation of diabetic kidney disease in Type 2 diabetes in south India. Diabetes Res Clin Pract 1999. 43(3):167-171. [DOD] [CrossRef]
- Vardarli I, Baier LJ, Hanson RL, Akkoyun I, Fischer C, Rohmeiss P, Basci A, Bartram CR, Van Der Woude FJ, Janssen B. Gene for susceptibility to diabetic nephropathy in type 2 diabetes maps to 18q22.3-23. Kidney Int 2002. 62(6):2176-2183. [DOD] [CrossRef]
- Bowden DW, Colicigno CJ, Langefeld CD, Sale MM, Williams A, Anderson PJ, Rich SS, Freedman BI. A genome scan for diabetic nephropathy in African Americans. Kidney Int 2004. 66(4):1517-1526. [DOD] [CrossRef]
- Knowler WC, Coresh J, Elston RC, Freedman BI, Iyengar SK, Kimmel PL, Olson JM, Plaetke R, Sedor JR, Seldin MF. The Family Investigation of Nephropathy and Diabetes (FIND): design and methods. J Diabetes Complications 2005. 19(1):1-9. [DOD] [CrossRef]
- Igo RP Jr, Iyengar SK, Nicholas SB, Goddard KA, Langefeld CD, Hanson RL, Duggirala R, Divers J, Abboud H, Adler SG, et al. Genomewide linkage scan for diabetic renal failure and albuminuria: the FIND study. Am J Nephrol 2011. 33(5):381-389. [DOD] [CrossRef]
- Freedman BI, Kopp JB, Winkler CA, Nelson GW, Rao DC, Eckfeldt JH, Leppert MF, Hicks PJ, Divers J, Langefeld CD, Hunt SC. Polymorphisms in the nonmuscle myosin heavy chain 9 gene (MYH9) are associated with albuminuria in hypertensive African Americans: the HyperGEN study. Am J Nephrol 2009. 29(6):626-632. [DOD] [CrossRef]
- Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 2010. 128(3):345-350. [DOD] [CrossRef]
- Iyengar SK, Abboud HE, Goddard KA, Saad MF, Adler SG, Arar NH, Bowden DW, Duggirala R, Elston RC, Hanson RL, et al. Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: the family investigation of nephropathy and diabetes (FIND). Diabetes 2007. 56(6):1577-1585. [DOD] [CrossRef]
- Alkayyali S, Lyssenko V. Genetics of diabetes complications. Mamm Genome 2014. 25(9-10):384-400. [DOD] [CrossRef]
- Williams WW, Salem RM, McKnight AJ, Sandholm N, Forsblom C, Taylor A, Guiducci C, McAteer JB, McKay GJ, Isakova T, et al. Association testing of previously reported variants in a large case-control meta-analysis of diabetic nephropathy. Diabetes 2012. 61(8):2187-2194. [DOD] [CrossRef]
- Song Y, Ailenberg M, Silverman M. Cloning of a novel gene in the human kidney homologous to rat munc13s: its potential role in diabetic nephropathy. Kidney int 1998. 53(6):1689-1695. [DOD] [CrossRef]
- Tregouet DA, Groop PH, McGinn S, Forsblom C, Hadjadj S, Marre M, Parving HH, Tarnow L, Telgmann R, Godefroy T, et al. G/T substitution in intron 1 of the UNC13B gene is associated with increased risk of nephropathy in patients with type 1 diabetes. Diabetes 2008. 57(10):2843-2850. [DOD] [CrossRef]
- Ma RC, Tam CH, Wang Y, Luk AO, Hu C, Yang X, Lam V, Chan AW, Ho JS, Chow CC, et al. Genetic variants of the protein kinase C-beta 1 gene and development of end-stage renal disease in patients with type 2 diabetes. JAMA 2010. 304(8):881-889. [DOD] [CrossRef]
- Wang F, Fang Q, Yu N, Zhao D, Zhang Y, Wang J, Wang Q, Zhou X, Cao X, Fan X. Association between genetic polymorphism of the angiotensin-converting enzyme and diabetic nephropathy: a meta-analysis comprising 26,580 subjects. J Renin Angiotensin Aldosterone Syst 2012. 13(1):161-174. [DOD] [CrossRef]
- Shi Z, Lu W, Xie G. Association between the RAGE gene -374T/A, -429T/C polymorphisms and diabetic nephropathy: a meta-analysis. Ren Fail 2015. 37(5):751-756. [DOD] [CrossRef]
- Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 1993. 329(27):2002-2012. [DOD] [CrossRef]
- Nagase S, Suzuki H, Wang Y, Kikuchi S, Hirayama A, Ueda A, Takada K, Oteki T, Obara M, Aoyagi K, Koyama A. Association of ecNOS gene polymorphisms with end stage renal diseases. Mol Cell Biochemi 2003. 244(1-2):113-118. [DOD] [CrossRef]
- Ma ZJ, Chen R, Ren HZ, Guo X, Chen JG, Chen LM. Endothelial nitric oxide synthase (eNOS) 4b/a polymorphism and the risk of diabetic nephropathy in type 2 diabetes mellitus: A meta-analysis. Meta Gene 2014. 2:50-62. [DOD] [CrossRef]
- Mooyaart AL, Valk EJ, van Es LA, Bruijn JA, de Heer E, Freedman BI, Dekkers OM, Baelde HJ. Genetic associations in diabetic nephropathy: a meta-analysis. Diabetologia 2011. 54(3):544-553. [DOD] [CrossRef]
- Hanson RL, Bogardus C, Duggan D, Kobes S, Knowlton M, Infante AM, Marovich L, Benitez D, Baier LJ, Knowler WC. A search for variants associated with young-onset type 2 diabetes in American Indians in a 100K genotyping array. Diabetes 2007. 56(12):3045-3052. [DOD] [CrossRef]
- Pezzolesi MG, Poznik GD, Mychaleckyj JC, Paterson AD, Barati MT, Klein JB, Ng DP, Placha G, Canani LH, Bochenski J, et al. Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes 2009. 58(6):1403-1410. [DOD] [CrossRef]
- McDonough CW, Palmer ND, Hicks PJ, Roh BH, An SS, Cooke JN, Hester JM, Wing MR, Bostrom MA, Rudock ME, et al. A genome-wide association study for diabetic nephropathy genes in African Americans. Kidney Int 2011. 79(5):563-572. [DOD] [CrossRef]
- Sandholm N, Salem RM, McKnight AJ, Brennan EP, Forsblom C, Isakova T, McKay GJ, Williams WW, Sadlier DM, Makinen VP, et al. New susceptibility loci associated with kidney disease in type 1 diabetes. Plos Genet 2012. 8(9):e1002921. [DOD] [CrossRef]
- Sandholm N, McKnight AJ, Salem RM, Brennan EP, Forsblom C, Harjutsalo V, Makinen VP, McKay GJ, Sadlier DM, Williams WW, et al. Chromosome 2q31.1 associates with ESRD in women with type 1 diabetes. J Am Soc Nephrol 2013. 24(10):1537-1543. [DOD] [CrossRef]
- Sambo F, Malovini A, Sandholm N, Stavarachi M, Forsblom C, Makinen VP, Harjutsalo V, Lithovius R, Gordin D, Parkkonen M, et al. Novel genetic susceptibility loci for diabetic end-stage renal disease identified through robust naive Bayes classification. Diabetologia 2014. 57(8):1611-1622. [DOD] [CrossRef]
- Yamada Y, Nishida T, Ichihara S, Kato K, Fujimaki T, Oguri M, Horibe H, Yoshida T, Watanabe S, Satoh K, et al. Identification of chromosome 3q28 and ALPK1 as susceptibility loci for chronic kidney disease in Japanese individuals by a genome-wide association study. J Med Genet 2013. 50(6):410-418. [DOD] [CrossRef]
- Sandholm N, Forsblom C, Makinen VP, McKnight AJ, Osterholm AM, He B, Harjutsalo V, Lithovius R, Gordin D, Parkkonen M, et al. Genome-wide association study of urinary albumin excretion rate in patients with type 1 diabetes. Diabetologia 2014. 57(6):1143-1153. [DOD] [CrossRef]
- Germain M, Pezzolesi MG, Sandholm N, McKnight AJ, Susztak K, Lajer M, Forsblom C, Marre M, Parving HH, Rossing P, et al. SORBS1 gene, a new candidate for diabetic nephropathy: results from a multi-stage genome-wide association study in patients with type 1 diabetes. Diabetologia 2015. 58(3):543-548. [DOD] [CrossRef]
- Cooke Bailey JN, Palmer ND, Ng MC, Bonomo JA, Hicks PJ, Hester JM, Langefeld CD, Freedman BI, Bowden DW. Analysis of coding variants identified from exome sequencing resources for association with diabetic and non-diabetic nephropathy in African Americans. Hum Genet 2014. 133(6):769-779. [DOD] [CrossRef]
- Veves A. Clinical management of diabetic neuropathy. Contemporary endocrinology. Humana Press, 1998, p. 337. [DOD]
- Gragnoli C. T2D-nephropathy linkage within 12q24 locus. Diabetes Res Clin Pract 2011. 92(3):e73-e75. [DOD] [CrossRef]
- Gragnoli C. Proteasome modulator 9 and macrovascular pathology of T2D. Cardiovasc Diabetol 2011. 10:32. [DOD] [CrossRef]
- Gragnoli C. Proteasome modulator 9 gene is linked to diabetic and non-diabetic retinopathy in T2D. Ophthal Genet 2011. 32(4):228-230. [DOD] [CrossRef]
- Gragnoli C. PSMD9 is linked to type 2 diabetes neuropathy. J Diabetes Complications 2011. 25(5):329-331. [DOD] [CrossRef]
- Zhang X, Sun Z, Jiang H, Song X. Relationship between single nucleotide polymorphisms in the 3'-untranslated region of the vascular endothelial growth factor gene and susceptibility to diabetic peripheral neuropathy in China. Arch Med Sci 2014. 10(5):1028-1034. [DOD] [CrossRef]
- Saraswathy R, Anand S, Kunnumpurath SK, Kurian RJ, Kaye AD, Vadivelu N. Chromosomal Aberrations and Exon 1 Mutation in the AKR1B1 Gene in Patients with Diabetic Neuropathy. Ochsner J 2014. 14(3):339-342. [DOD]
- Wu S, Han Y, Hu Q, Zhang XJ, Cui GC, Li ZZ, Guan YT. Effects of common polymorphisms in the MTHFR and ACE genes on diabetic peripheral neuropathy progression: a meta-analysis. Mol Neurobiol 2014. In press. [DOD]
- Li Y, Tong N. Angiotensin-converting enzyme I/D polymorphism and diabetic peripheral neuropathy in type 2 diabetes mellitus: A meta-analysis. J Renin Angiotensin Aldosterone Syst 2014. In press. [DOD]
- Tang TS, Prior SL, Li KW, Ireland HA, Bain SC, Hurel SJ, Cooper JA, Humphries SE, Stephens JW. Association between the rs1050450 glutathione peroxidase-1 (C > T) gene variant and peripheral neuropathy in two independent samples of subjects with diabetes mellitus. Nutr Metab Cardiovasc Dis 2012. 22(5):417-425. [DOD] [CrossRef]
- Ciccacci C, Morganti R, Di Fusco D, D'Amato C, Cacciotti L, Greco C, Rufini S, Novelli G, Sangiuolo F, Marfia GA, et al. Common polymorphisms in MIR146a, MIR128a and MIR27a genes contribute to neuropathy susceptibility in type 2 diabetes. Acta Diabetol 2014. 51(4):663-671. [DOD] [CrossRef]
- Meng W, Deshmukh HA, van Zuydam NR, Liu Y, Donnelly LA, Zhou K, Morris AD, Colhoun HM, Palmer CN, Smith BH. A genome-wide association study suggests an association of Chr8p21.3 (GFRA2) with diabetic neuropathic pain. Europ J Pain 2015. 19(3):392-399. [DOD] [CrossRef]
- Yuan H, Reddy MA, Sun G, Lanting L, Wang M, Kato M, Natarajan R. Involvement of p300/CBP and epigenetic histone acetylation in TGF-beta1-mediated gene transcription in mesangial cells. Am J Physiol Renal Physiol 2013. 304(5):F601-F613. [DOD] [CrossRef]
- Reddy MA, Tak Park J, Natarajan R. Epigenetic modifications in the pathogenesis of diabetic nephropathy. Sem Nephrol 2013. 33(4):341-353. [DOD] [CrossRef]
- Reddy MA, Sumanth P, Lanting L, Yuan H, Wang M, Mar D, Alpers CE, Bomsztyk K, Natarajan R. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int 2014. 85(2):362-373. [DOD] [CrossRef]
- Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015. 58(3):443-455. [DOD] [CrossRef]
- Syreeni A, El-Osta A, Forsblom C, Sandholm N, Parkkonen M, Tarnow L, Parving HH, McKnight AJ, Maxwell AP, Cooper ME, Groop PH. Genetic examination of SETD7 and SUV39H1/H2 methyltransferases and the risk of diabetes complications in patients with type 1 diabetes. Diabetes 2011. 60(11):3073-3080. [DOD] [CrossRef]
- Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, Ziemann M, Karagiannis T, Tonna S, Kowalczyk A, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 2011. 21(10):1601-1615. [DOD] [CrossRef]
- Kim ES, Isoda F, Kurland I, Mobbs CV. Glucose-induced metabolic memory in Schwann cells: prevention by PPAR agonists. Endocrinology 2013. 154(9):3054-3066. [DOD] [CrossRef]
- McKnight A, Swan EJ, Patterson CC, Maxwell AP. Changes in DNA methylation are associated with diabetic nephropathy in individuals with type 1 diabetes. The Diabetes UK Professional Conference, Liverpool, March 3-7, 2014. [DOD]
- Swan EJ, Maxwell AP, McKnight AJ. Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood-derived DNA from individuals with Type 1 diabetes. Diabet Med 2015. In press. [DOD]
- Zeng J, Chen B. Epigenetic mechanisms in the pathogenesis of diabetic retinopathy. Ophthalmologica 2014. 232(1):1-9. [DOD] [CrossRef]
- McArthur K, Feng B, Wu Y, Chen S, Chakrabarti S. MicroRNA-200b regulates vascular endothelial growth factor-mediated alterations in diabetic retinopathy. Diabetes 2011. 60(4):1314-1323. [DOD] [CrossRef]
- Silva VA, Polesskaya A, Sousa TA, Correa VM, Andre ND, Reis RI, Kettelhut IC, Harel-Bellan A, De Lucca FL. Expression and cellular localization of microRNA-29b and RAX, an activator of the RNA-dependent protein kinase (PKR), in the retina of streptozotocin-induced diabetic rats. Molecul Vis 2011. 17:2228-2240. [DOD]
- Kato M, Arce L, Natarajan R. MicroRNAs and their role in progressive kidney diseases. Clin J Am Soc Nephrol 2009. 4(7):1255-1266. [DOD] [CrossRef]
- Wang Q, Wang Y, Minto AW, Wang J, Shi Q, Li X, Quigg RJ. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J 2008. 22(12):4126-4135. [DOD] [CrossRef]
- Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 2008. 83(3):311-321. [DOD] [CrossRef]
- Lin Z, Huang G, Zhang J, Lin X. Adiponectin gene polymorphisms and susceptibility to diabetic nephropathy: a meta-analysis. Ren Fail 2014. 36(3):478-487. [DOD] [CrossRef]
- Cho H, Sobrin L. Genetics of diabetic retinopathy. Curr Diabetes Rep 2014. 14(8):515. [DOD] [CrossRef]
- Palmer ND, Ng MC, Hicks PJ, Mudgal P, Langefeld CD, Freedman BI, Bowden DW. Evaluation of candidate nephropathy susceptibility genes in a genome-wide association study of African American diabetic kidney disease. Plos One 2014. 9(2):e88273. [DOD] [CrossRef]
|


)
)

