Chapter I. Pathogenesis and Function
| Rev Diabet Stud,
2015,
12(3-4):260-276 |
DOI 10.1900/RDS.2015.12.260 |
Beyond the Protein-Coding Sequence: Noncoding RNAs in the Pathogenesis of Type 2 Diabetes
Johanna K. DiStefano
Translational Genomics Research Institute, Phoenix, AZ 85004, USA
Manuscript submitted February 24, 2015; resubmitted April 1, 2015; accepted April 2, 2015.
Keywords: type 2 diabetes, noncoding RNA, microRNA, miRNA-binding site, lncRNA, epigenetics, beta-cell, insulin resistance, genetic variant, SNP
Abstract
Diabetes mellitus results from a deficiency or failure to maintain normal glucose homeostasis. The most common form of the disease is type 2 diabetes (T2D), a progressive metabolic disorder characterized by elevated glucose levels that develops in response to either multi-organ insulin resistance or insufficient insulin secretion from pancreatic β-cells. Although the etiology of T2D is complex, many factors are known to contribute to defects of glucose homeostasis, including obesity, unhealthy lifestyle choices, genetic susceptibility, and environmental exposures. In addition to these factors, noncoding RNAs (ncRNAs) have been recently implicated in the pathogenesis of T2D, playing roles in several of the pathophysiological mechanisms underlying the disease, particularly in insulin-sensitive tissues such as pancreatic β-cells, liver, muscle, and adipose tissue. A growing number of publications demonstrate that polymorphisms in ncRNAs or their target genes may represent a new class of genetic variation contributing to the development of T2D. This review summarizes both the current state of knowledge of ncRNAs, specifically microRNAs (miRNAs), involved in the regulation of β-cell function, insulin sensitivity, and insulin action in peripheral organs. The role of genetic variation in miRNAs or miRNA binding sites in the pathogenesis of T2D is also discussed. While far less is known about the impact of long ncRNAs (lncRNAs) in the development of T2D, emerging evidence suggests that these molecules may be able to contribute to β-cell dysfunction in response to hyperglycemia. This article provides an overview of the studies conducted to date in this field, focusing on lncRNAs that are dysregulated in human pancreatic islets.
Abbreviations: AKT2 – v-akt murine thymoma viral oncogene homolog 2; Akt3 – thymoma viral proto-oncogene 3; ANRIL – cyclin-dependent kinase inhibitor 2B antisense RNA 1; AP3S2 – adaptor-related protein complex 3, sigma 2 subunit; Basp1 – brain-abundant, membrane-attached signal protein 1; BKS – Black Kaliss J; cMaf – avian musculoaponeurotic fibrosarcoma oncogene homolog; Cplx1 – complexin 1; Cspa – RNA chaperone and antiterminator, cold-inducible; DIAGRAM – Diabetes Genetics Replication and Meta-Analysis; DLK1 – delta-like 1 homolog; ER – endoplasmic reticulum; eQTL – expression quantitative trait locus; eRNA – enhancer RNA; Flot2 – flotillin 2; Foxa2 – forkhead box A2; GK – Goto-Kakizaki; GLI – glioma-associated oncogene; GLIS3 – GLI-similar zinc finger 3; GLUT4 – glucose transporter type 4; HbA1c – glycated hemoglobin; HIPK3 – homeodomain interacting protein kinase 3; Hnf1a – hepatic nuclear factor 1 homeobox A; HOMA – homeostatic model assessment; IAPP – islet amyloid polypeptide; IGF1R – insulin-like growth factor 1; INSR – insulin receptor; IRS2 – insulin receptor substrate 2; KCNK16 – potassium two pore domain channel, subfamily K, member 16; KCNQ1OT1 – potassium voltage-gated channel, subfamily Q, member 1, opposite strand/antisense transcript 1 (non-protein coding); Kir6.2 – potassium inwardly rectifying channel, subfamily J, member 11; lincRNA – long intervening noncoding RNA; lncRNA – long non-coding RNA; MADD – MAP-kinase activating death domain; MCT1 – membrane monocarboxylate transporter; MEF2C – myocyte enhancer factor 2C; MEG3 – maternally expressed 3; MIN6 – mouse insulinoma 6 cells; miRNA – microRNA; MODY – maturity-onset diabetes of the young; MTOR – mechanistic target of rapamycin; MTPN – myotrophin; NAT – natural antisense transcripts; ncRNA – noncoding RNA; NeuroD1 – neurogenic differentiation 1; NOTCH2 – notch 2; OneCut2 – one cut domain, family member 2; ORP8 – oxysterol-binding protein-related protein 8; paRNA – promoter-associated RNA; PAX6 – paired box 6; PDK1 – 3'-phosphoinositide-dependent protein kinase-1; pdx-1 – pancreatic and duodenal homeobox 1; PEPCK – phosphoenolpyruvate carboxykinase gene; Pfn2 – profilin 2; PI3K – p85 subunit of phosphatidylinositol 3-kinase; PIK3IP1 – phosphoinositide-3-kinase interacting protein 1; piRNA – Piwi-interacting RNA; Pkcb – protein kinase C, beta; RICTOR – RPTOR-independent companion of the mechanistic target of rapamycin, complex 2; RPTOR – regulatory-associated protein of MTOR complex 1; siRNA – small interfering RNA; Sirt1 – sirtuin 1; Slc25a22 – solute carrier, family 25, member 22; SLC30A8 – solute carrier, family 30, member 8; SNAP – soluble N-ethylmaleimide-sensitive factor attachment protein; SNARE – SNAP receptor; Snca – synuclein alpha; sQTL – splicing quantitative trait locus; Sur1 – ATP-binding cassette, sub-family C (CFTR/MRP), member 8; SYT11 – synaptotagmin 11; T2D – type 2 diabetes; TP53INP1 - p53-induced nuclear protein 1; TSC1 – tuberous sclerosis 1; TXNIP – thioredoxin-interacting protein; VPS26A – vacuolar protein sorting-associated protein 26 retromer complex, component A; WFS1 – wolframin ER transmembrane glycoprotein; Wipf2 – Wiskott-Aldrich syndrome-like interacting protein family, member 2; Zdhhc – zinc finger type, DHHC-containing
1. Introduction
Diabetes mellitus, a multifactorial, heterogeneous collection of disorders, results from a deficiency or failure to maintain normal glucose homeostasis. Type 2 diabetes (T2D) develops in response to multi-organ insulin resistance and/or inadequate insulin secretion from pancreatic β-cells [1, 2]. Due to a combination of different factors, including increased levels of sedentary behavior and easy access to high-energy foods, the prevalence of T2D has risen sharply in recent decades, presently affecting over 300 million individuals worldwide [3]. The predicted trajectory of diabetes is grim, with current estimates of the number of people affected with the disease exceeding 520 million within fifteen years [4]. At this time, T2D is the eighth leading cause of death worldwide and accounts for approximately 2.7% of all deaths [5]. For most industrialized countries, T2D represents a major public health concern.
The etiology of T2D remains incompletely understood, although insulin is recognized as the key mediator of disease development [1, 2]. The pathogenesis of the disease is complex and multifactorial, and genetic predisposition, environmental exposures, and lifestyle factors mediate susceptibility to T2D [6]. However, while obesity, increased levels of sedentary behavior, and aging are known to contribute to an elevated risk of developing T2D, there is also a substantial proportion of people who do not develop the disease in the face of these exposures. In addition, while genome-wide association studies have identified a number of polymorphisms, together they account only for a small proportion of the inter-individual variation in disease susceptibility [7-10]. Notably, nearly 90% of the variants identified to date map to non-protein coding regions, a finding which challenges our understanding of the functional impact of such genetic factors [11-13]. Despite intense research efforts to identify specific biological targets and signaling pathways relative to these factors, the molecular mechanisms by which environmental influences affect the pathogenesis of T2D in susceptible individuals remain largely unknown. Growing evidence implicates members of a relatively new class of nucleic acids, non-coding RNAs (ncRNAs), in the etiology of T2D and, given the expression patterns and function of these molecules, as discussed below, ncRNAs may also represent potential mediators of environmental influences on pathological mechanisms underpinning the disease [14].
Approximately 90% of the human genome is transcribed [11]. In contrast to the central dogma underlying the transmission of genetic information from DNA to RNA to protein, only a small percentage of transcripts in the human transcriptome give rise to proteins, Indeed, most of the human transcriptome is transcribed as non-coding RNA (ncRNA), that is, RNA that does not encode protein [11, 13]. ncRNA can be broadly classified into two classes: infrastructural and regulatory. Infrastructural ncRNA molecules are involved in mRNA translation (ribosomal RNA and transfer RNA), as well as splicing and rRNA modification (small nuclear RNA and small nucleolar RNA, respectively). In contrast, regulatory ncRNAs mainly regulate gene expression and consist of a repertoire of different species including microRNAs (miRNAs), Piwi-interacting RNAs (pi-RNAs), small interfering RNAs (siRNAs), promoter-associated RNAs (paRNAs), enhancer RNAs (eRNAs), and long non-coding RNAs (lncRNAs) [15, 16].
Regulatory ncRNAs are commonly characterized by transcript size, but there are additional differences in both biological function and molecular/cellular effects that serve to distinguish among them. The best-characterized members of the small regulatory ncRNAs are miRNAs, which are endogenous, single-stranded RNAs (21-25 nucleotides in length) that regulate gene expression by blocking translation or promoting cleavage of specific target mRNAs, or less frequently, by targeting the promoter region [17]. miRNAs exert effects through specific base pair recognition mechanisms to modulate expression of target genes [18, 19] and approximately 20-30% of all human genes are transcriptionally regulated by miRNAs via targeting sequences in the 3’ untranslated region (3'-UTR) [20, 21]. Aberrant regulation of miRNAs has been associated with the development of many diseases including neurologic disorders, cardiovascular diseases, and certain cancers [22, 23]. miRNAs have also been found to be critical for pancreatic development [24-26], maintenance of β-cell identity [27], and regulation of biological processes related to T2D pathogenesis, including glucose-stimulated insulin secretion, glucose uptake in muscle and liver, adipogenesis, and adipocyte function [28-30].
Until recently, investigations of genetic factors leading to disease susceptibility have primarily focused on the study of protein-coding sequences. However, a growing number of reports indicates that genetic variation in miRNA genes may be involved in the development of disease either through the regulation of miRNA expression or via alteration of miRNA binding sites within mRNA regulatory regions. Recent studies have demonstrated that:
1. Predicted mRNA targets of islet-expressed miRNAs are globally enriched for signals of T2D association [31].
2. Predicted mRNA target sites for islet-expressed miRNAs overlap with potentially causal variants in loci showing genome-wide evidence for association with T2D [31].
3. Polymorphisms in miRNAs are associated with T2D susceptibility [32].
Thus, the investigation of polymorphisms in miRNAs and miRNA targets promises new insight into the complex natures of both miRNA expression and function and genetic predisposition to T2D; it also expands our understanding of the pathophysiology of the disease.
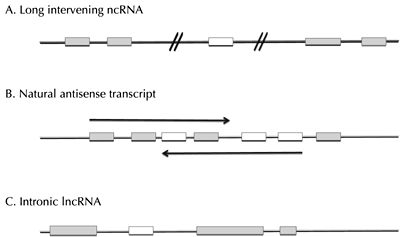
The second class of ncRNAs, long ncRNAs (lncRNAs), is comprised of transcribed RNA molecules with much longer lengths than small ncRNAs, typically >200 nucleotides. To date, three categories of lncRNAs have been identified (Figure 1), including long intervening ncRNAs (lincRNAs), which are transcribed from regions at least >1kb from protein-coding genes; natural antisense transcripts (NAT), which are transcribed from the opposite strand of a protein-coding gene; and intronic lncRNAs, which are transcribed from introns of protein-coding genes [33, 34]. lncRNAs are post-transcriptionally processed like mRNAs but do not result in protein synthesis [35-37]. In general, lncRNAs bind to DNA, RNA, and protein and exert effects through roles that include directing chromatin-modifying complexes to specific genomic loci, providing molecular scaffolds, modulating transcriptional programs, and regulating miRNA expression [34]. Thus far, lncRNAs have been shown to play a role in the regulation of gene expression, genomic imprinting, maintenance of pluripotency, nuclear organization and compartmentalization, and alternative splicing [34, 37-39]. Like miRNAs, lncRNAs have been found to be dysregulated in human diseases, although the role these molecules play in the disease process is not understood nearly as well [40, 41]. However, although far less is known about the impact of lncRNAs in the development of T2D, there is growing evidence that these molecules also contribute to β-cell dysfunction in response to hyperglycemia.
 |
 |
Figure 1. The three main categories of lncRNAs. A. lincRNAs, B. NATs, and C. intronic RNAs. Grey boxes represent protein-coding genes, with transcriptional direction depicted by arrows. Unfilled boxes depict lncRNAs. Diagonal lines in lincRNAs represent distances >1 kb. Adapted from [34]. |
|
The role of ncRNAs in the pathogenesis of T2D has only recently become recognized, but there is already convincing support for their involvement in many of the pathophysiological mechanisms underlying the disease. The goal of this review article is to summarize the current state of knowledge of miRNAs that have been implicated in both β-cell function and regulation of insulin sensitivity or insulin action. This review also addresses the role of genetic variation in either miRNAs or miRNA binding sites within target mRNAs in the pathogenesis of T2D. Finally, an overview of the studies conducted to date is provided, with a focus on lncRNAs that are dysregulated in human pancreatic islets.
2. The role of miRNAs in T2D pathogenesis
2.1 miRNAs involved in the regulation of pancreatic β-cell function
Pancreatic β-cells are specialized endocrine cells that synthesize and secrete insulin in response to glucose stimulation. When β-cell functioning is compromised, typically through autoimmune destruction, chronic metabolic derangement, or exposure to environmental factors, insulin secretion decreases, glucose homeostasis becomes impaired, and diabetes mellitus may eventually develop. In addition to genetic and environmental influences, miRNAs have been shown to regulate biological processes in the β-cell, including β-cell differentiation and proliferation, as well as insulin biosynthesis and secretion. These miRNAs and their mechanisms of action have been extensively reviewed elsewhere [28, 42-44]. In this review, we focus mostly on recent findings in the field of miRNAs involved in the regulation of β-cell function relative to insulin resistance and T2D (Table 1).
Table
1.
miRNAs implicated in T2D pathogenesis |
|
 |
|
To investigate the overall importance of miRNAs in β-cells, Kalis et al. generated mice with a β-cell-specific disruption of Dicer1 [24], an endoribonuclease that plays a critical role in the processing and generation of miRNAs [45]. Dicer1-depleted mice exhibited reduced insulin gene expression and impaired insulin secretion, both of which preceded the development of progressive hyperglycemia and diabetes. These animals also showed altered islet cell morphology, reduced β-cell mass, and differential pancreatic islet morphology, although fetal and neonatal β-cell development and insulin secretory function were normal [24, 26]. Thus, despite normal β-cell development, disruption of the miRNA network effectively resulted in defective insulin secretion, hyperglycemia, and diabetes, implying a significant function for miRNAs in this cell type. In another study, Yan and colleagues observed that the tissue-specific diurnal expression of Dicer1 was attenuated by diabetes [46]. Loss of Dicer1 rhythmicity corresponded with altered circadian expression patterns of specific miRNAs, including miR-146a and miR-125a-5p, indicating that diabetes produces effects on Dicer-controlled miRNAs. These results also suggest that restoring Dicer1 activity may potentially reverse some of the deleterious consequences of diabetes on specific miRNAs. Additional studies are necessary to address this intriguing possibility.
The role of miR-375 in pancreatic β-cell development and function has been well established. In islets, overexpression of miR-375 inhibits glucose-induced insulin secretion, while suppression of its function results in increased levels of insulin secretion [47]. One of the first validated targets of miR-375 was myotrophin (MTPN), and its inhibition produced effects on glucose-stimulated insulin secretion and release comparable to miR-375. Levels of PDK1 (3'-phosphoinositide-dependent protein kinase-1), another target of miR-375, increased in response to glucose treatment concomitantly with reduced miR-375 [48]. Mice with disrupted miR-375 expression (i.e., 375KO) exhibited hyperglycemia, increased levels of gluconeogenesis and hepatic glucose output, and higher levels of fasting and fed glucagon [49]. In these animals, impaired β-cell proliferation corresponded with decreased β-cell mass. In ob/ob mice, a model of increased β-cell mass and obesity-related diabetes, islet miR-375 expression was increased [49], while in fed diabetic Goto-Kakizaki (GK) rats, a model for non-obese insulin resistance and T2D, islet miR-375 expression was reduced [48]. Deletion of miR-375 in ob/ob mice (375/ob) led to a severe reduction in the proliferative capacity of the endocrine pancreas, resulting in diabetes [49]. Interestingly, targeted knockdown of miR-375 in zebrafish corresponded with aberrant migration of pancreatic islet cells and malformation of the endocrine pancreas [50]. In summary, the findings from animal studies provide compelling evidence that miR-375 plays an important role in β-cell function, and indicate that dysregulation of this miRNA may contribute significantly to the development of diabetes.
In humans, several studies have shown that miR-375 is upregulated in the pancreas or plasma of patients with T2D compared to non-diabetic individuals [51-53], while others found no evidence for differential expression [54] or observed decreased expression levels in individuals with impaired glucose tolerance [55]. Altered miR-375 expression has been correlated with changes in methylation status of its promoter [51, 52, 55]. Increased pancreatic miR-375 expression was associated with islet amyloid deposition, decreased β-cell mass, and reduced islet mitochondrial density, but the mechanism by which this miRNA contributes to these manifestations is not yet known [53]. In miRNA profiling analyses of primary human islets and enriched β-cell preparations, discussed in detail below, high expression of miR-375 was observed, predominantly in β-cells [31]. Together, the results reported thus far are consistent with a role for miR-375 in glucose homeostasis and pancreatic β-cell expansion in response to the increasing insulin demands associated with insulin resistance and T2D in humans [56].
Latreille et al. recently performed a comprehensive assessment of miR-7, a highly conserved, prototypical neuroendocrine miRNA [57]. Levels of miR-7 were 15-fold higher in pancreatic islets compared to adrenal glands, and of the miR-7 precursors, miR-7a2 was the most abundantly expressed in islets. To investigate the role of miR-7a in pancreatic β-cells, the researchers generated Mir7a2 conditional knockout mice and found that miR-7a2 deletion in β-cells improved glucose tolerance by increasing insulin secretion. miR-7a2 was found to regulate β-cell function by directly regulating genes such as Snca, Cspa, Cplx1, Pkcb, Basp1, Pfn2, Wipf2, and Zdhhc that control late stages of insulin granule fusion with the plasma membrane and ternary SNARE complex activity. Levels of miR-7a were significantly decreased in compensating islets of high fat diet-fed and ob/ob mice, as well as normoglycemic and compensating BKS db/db mice, but were increased in decompensating BKS db/db islets. To demonstrate the pathological significance of increased miR-7a levels in pancreatic islets of diabetic BKS db/db mice, transgenic mice overexpressing miR-7a in β-cells were generated. These animals developed hyperglycemia, reduced plasma insulin levels, and impaired glucose-stimulated insulin secretion, but showed no changes in β-cell proliferation or apoptosis. Together, these findings established miR-7 as the first negative regulator of insulin secretion in β-cells through a network regulating insulin granule exocytosis in pancreatic β-cells.
In addition to miR-375 and miR-7, miR-124a and miR-184 have been shown to affect β-cell function. In pancreatic islets from T2D donors, miR-124a was expressed at much higher levels compared to normal islets [54]. Overexpression of miR-124a in MIN6 cells resulted in impaired glucose-stimulated insulin secretion and silencing of this miRNA correlated with increased expression of predicted target genes of relevance to β-cell function, including Mtpn, Foxa2, Flot2, Akt3, Sirt1, and NeuroD1. Preliminary functional analyses showed that miR-124a bound to the 3'UTRs of both Mtpn and Foxa2. Likewise, in MIN6 cells overexpressing or underexpressing miR-124a, levels of Foxa2 were reduced or elevated, respectively [58]. A correlation between Foxa2 protein levels and decreased expression of pdx-1, Kir6.2, Sur-1, and Preproinsulin, all of which are involved with β-cell function, insulin secretion, and glucose metabolism, revealed a potential set of pathways by which miR-124a might contribute to diabetes pathogenesis through regulation of its target mRNA [58]. In a different signaling cascade, overexpression of miR-124a was found to correspond with decreased Foxa2 expression and reduced binding to the islet amyloid polypeptide (IAPP) promoter, resulting in lower levels of IAPP mRNA and protein expression [59]. In addition, miR-124a overexpression also inhibited induction of IAPP expression by thioredoxin-interacting protein (TXNIP), which is known to play a crucial role in glucotoxicity. Thus, the miR-124a/TXNIP/Foxa2/IAPP signaling cascade may link the TXNIP and IAPP pathways in the β-cell.
The expression of miR-184, which is highly enriched in pancreatic islets, has been negatively correlated with insulin secretion [60] and significant inhibition of glucose-stimulated insulin secretion in MIN6 cells [61] via mechanisms involving suppression of its target, Slc25a22. miR-184 showed expression changes in two different mouse models of obesity-induced diabetes prior to the onset of disease, but was also found to have positive effects on β-cell activity and mass [62]. Another study found that miR-29a/b directly binds the 3'UTR of the membrane monocarboxylate transporter (MCT1) in both human and mouse β-cells [63]. The two miRNAs were both found to target endogenous Mct1, leading to decreased protein expression. Inhibition of miR-29a in primary mouse islets corresponded to increased Mct1 mRNA levels, demonstrating that miR-29 isoforms contribute to silencing of MCT1 in β-cells, which may affect insulin release.
Other miRNAs with potential effects in pancreatic islets have been identified. For example, overexpression of miR-24 was correlated with β-cell proliferation and reduced insulin secretion in islets from db/db mice fed a high-fat diet [64]. Targets of miR-24 included Hnf1a and neurod1, both of which are known to cause maturity-onset diabetes of the young (MODY). Inhibition of either Hnf1a or neurod1 mimicked the effects of miR-24 overexpression, suggesting that miR-24 and specific MODY genes represent a novel pathway that may contribute to the pathogenesis of T2D, possibly through mechanisms involving over-nutrition. Expression of another miRNA, miR-187, was found to be approximately seven-fold more abundant in pancreatic islets from T2D patients compared to healthy controls, and was inversely correlated with glucose-induced insulin secretion in normoglycemic individuals [65]. Overexpression of this miRNA also decreased glucose-induced insulin secretion in primary cultures of rat islets and INS-1 cells, without affecting either insulin content or apoptotic index [65]. Homeodomain interacting protein kinase 3 (HIPK3), which regulates insulin secretion and glucose tolerance [66], was identified and validated as a direct miR-187 target, and levels of HIPK3 were reduced in islets from patients with T2D, suggesting that miR-187 may exert effects on glucose-stimulated insulin secretion through mechanisms involving HIPK3 [65]. Evidence for another miRNA, miR-9, demonstrated an effect on glucose-induced insulin secretion through the inhibition of OneCut2 expression in β-cells [67]. Elevated miR-9 also diminished sirt1 expression in β-cells [68]. Additional functional studies are expected to further clarify the roles of all of these miRNA/mRNA relationships in the pathogenesis of T2D.
In addition to single miRNAs, networks of miRNA may also contribute to glucose homeostasis. For example, transgenic mice with global and pancreas-specific Cre-inducible activation of Let-7a, Let-7d, and Let-7f expression exhibited reduced glucose-stimulated insulin secretion, resulting in impaired glucose tolerance [69]. In contrast, global knockdown of the Let-7 family members prevented obesity-induced glucose intolerance and restored insulin signaling in muscle and liver. Together, the results demonstrate that Let-7 influences different aspects of glucose metabolism in multiple tissues, and that inhibition of Let-7 may improve pancreatic β-cell function. However, the precise role that this family of miRNAs plays in the pathogenesis of the disease remains to be determined.
2.2 miRNA profiling of pancreatic islets and β-cells
A number of studies have recently applied miRNA profiling approaches in pancreatic islets or β-cells from diabetic and non-diabetic donors to gain new insight into the molecular mechanisms underlying T2D. A comprehensive investigation by van de Bunt and colleagues [31] utilized next generation sequencing to profile the miRNA fraction in primary human islets and preparations of β-cells enriched by fluorescence-activated cell sorting to purity levels >90%. The authors identified 366 and 346 miRNAs that were expressed in islets and enriched β-cells, respectively, including a total of 384 unique miRNAs, of which 328 were shared between islets and β-cells. The miRNA expression profiles in islets and β-cells were strongly correlated (r2 = 0.78), although miR-375 was present at significantly higher levels in β-cells compared to islets (42% vs. 27%). In contrast, levels of miR-143-3p were much lower in β-cells (2% vs. 16%). Some of the most highly expressed miRNAs identified in islets were those that had been previously identified in other studies, including miR-375, miR-7-5p, miR-148a-3p, miR-26a-5p, and miR-127-3p [31], while others, such as miR-27b-3p and miR-192-5p, represented novel findings. In comparisons with publically available miRNA expression data for over fifteen different human tissues, the authors determined that 40 of the 366 miRNAs identified were islet-specific. Several of the ten most tissue-specific miRNAs have been previously implicated in islet function (miR-184, miR-375, miR-182-5p, and miR-127-5p), while others (miR-409-5p, miR-1468, miR-183-5p, miR-136-3p, miR-370, and miR-153) represent potentially novel players in the maintenance of islet identity.
Klein et al. also assessed miRNA profiles in enriched preparations of pancreatic α- and β-cells from six human pancreases using a microarray approach [70]. The majority of miRNAs were preferentially expressed in β-cells: out of the 141 miRNAs identified in the analyses, only seven were expressed in α-cells. Several of the identified β-cell-specific miRNAs have been previously associated with the regulation of insulin expression and content and/or overlapped with the findings from van de Bunt et al. including miR-204, miR-127-3p, miR-184, miR-148, miR-29, miR-375, and miR-7 [31]. The transcription factor, cMaf, which is expressed exclusively in α-cells where it regulates expression of the glucagon gene, was identified as a target for several β-cell-specific miRNAs, including miR-125b, miR-182, and miR-200c, which showed 27.3-, 9.7-, and 3.3-fold higher expression in β-cells compared to α-cells [70]. Inhibition of these three miRNAs in MIN6 cells resulted in increased cMaf expression, while their overexpression in αTC6 cells corresponded with decreased cMaf expression.
The work of van de Bunt et al. [31] and Klein et al. [70] was performed using islets from donors without a known T2D diagnosis. In addition to these investigations, a small number of profiling studies have been performed using islets from diabetic donors. For example, a recent microarray analysis to profile miRNAs in pancreatic islets of prediabetic and diabetic db/db mice and mice fed a high-fat diet identified two distinct categories of differentially expressed molecules [62]. miR-132, miR-184, and miR-338-3p exhibited expression changes in islets well before the onset of diabetes, while miR-34a, miR-146a, miR-199a-3p, miR-203, miR-210, and miR-383 showed differences mostly in diabetic mice. Expression changes in prediabetic animals exerted positive effects on β-cell activity and mass, and those in diabetic mice increased β-cell apoptosis. These findings suggest that obesity and insulin resistance produce changes in miRNAs, with initially sustained β-cell function, but β-cell loss and development of T2D after further deregulation in additional miRNAs [62]. These results also indicated that the maintenance of glucose homeostasis, or in contrast, the development of glucose intolerance, might be mediated by alterations in expression patterns of specific miRNAs.
In a global profiling assessment of 384 miRNAs in pancreatic islets from non-obese T2D GK and non-diabetic Wistar rats, Esquerra et al. identified 30 differentially expressed miRNAs, most of which were upregulated in diabetic islets [71]. Glucose-induced insulin secretion experiments using isolated islets from GK and control animals revealed significant differences in the magnitude and direction of miRNA expression, suggesting differences in short- and long-term glucose dependence. In GK rats, expression of miR-130a, miR-132, miR-212, and miR-335 was regulated by hyperglycemia and the putative targets of these upregulated miRNAs were enriched for insulin secretion-related genes. The authors hypothesized that the insulin secretion defects in the GK rat may be due, in part, to upregulation of miRNAs that regulate key proteins involved in insulin exocytosis from β-cells. In support of these findings, glucose regulation of miR-132 and miR-212 were also reported in MIN6 cells [72], and these two miRNAs were upregulated in pancreatic islets in obese mice [73], suggesting a common pathway for these ncRNAs in disease pathogenesis for both diabetic GK rats and obese mice models.
In humans, Bolmeson et al. compared global miRNA expression patterns in islets from nine healthy donors and six glucose intolerant individuals using a bead-based technology [60]. A total of 319 miRNAs were measured, and of these, miR-375, miR-127-3p, miR-184, miR-195, and miR-493 showed enrichment in pancreatic islets compared to liver and skeletal muscle. In islets from glucose intolerant donors, levels of miR-21, miR-375, and miR-127-3p were increased compared to normal islets and expression of miR-375, miR-122, miR-184, and miR-127-3p correlated positively with insulin gene expression, indicating that some of these miRNAs may be exerting effects on glucose homeostasis through insulin synthesis. A similar study assessed miRNA content using TaqMan arrays in islets from 11 T2D donors and nine controls [65]. Of the 667 miRNA assays on the array, 255 amplified in all samples. Of these, only miR-187 and miR-345 showed statistically significant differential expression between islets from diabetic donors and unaffected controls. In an independent sample of islets from ten individuals with T2D and ten without the disease, the authors continued to observe significantly higher levels of miR-187, but not miR-345. Levels of miR-187 were also inversely correlated with glucose-stimulated insulin secretion in a sample of 35 normoglycemic donors [65]. In earlier studies of the islet miRNome conducted in donors without known T2D [31, 70], miR-187 levels were low or undetectable, suggesting that expression of this miRNA corresponds with the deteriorating condition of the β-cell during the progressive development of T2D.
Most recently, Kameswaran and colleagues sequenced small RNAs from islets of three nondiabetic and four T2D donors [74]. A total of 15 (out of 800) miRNAs showed significantly different expression between T2D and nondiabetic donors, some of which shared overlap with earlier studies, including miR-487-3p, miR-495-3p, miR-539-3p, miR-369-3p, miR-7-3-5p, miR-30a-5p, miR-7-1-3p, miR-656-3p, miR-487a-3p, miR-23c, miR-4716-3p, miR-544a-5p, miR-589-5p, miR-216a-5p, and miR-187-3p. Out of the top ten downregulated miRNAs, seven were derived from the maternally expressed, imprinted DLK1-MEG3 locus on 14q32. As seen in earlier studies [70, 75], expression levels of the 14q32 miRNAs and the long noncoding MEG3 RNA were ~16-20-fold higher in β-cells compared to α-cells. Potential targets of the 14q32 miRNAs included islet amyloid polypeptide (IAPP) and p53-induced nuclear protein 1 (TP53INP1), both of which increase β-cell apoptosis when overexpressed in islets. A direct targeting relationship was observed between miR-376a and miR-432 and the IAPP 3'UTR. TP53INP1 transcript levels were increased in islets from donors with T2D compared to non-diabetic individuals and miR-495 was found to downregulate TP53INP1 expression.
Unlike investigations of circulating miRNAs as potential markers for T2D where there is minimal overlap among reported findings [76-85], miRNA profiling studies in islets and β-cells have shown strongly consistent results (Table 2). At this time, however, little is known of the potential targets of these miRNAs and the mechanisms by which they affect disease development in susceptible individuals. Additional studies aimed at delineating specific miRNA/mRNA networks and consequent effects on β-cell function are expected to further enhance our understanding of the complex pathogenesis of T2D.
Table
2.
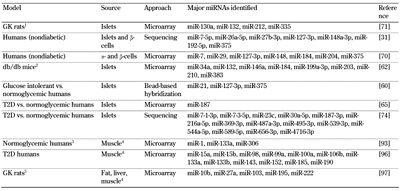
Major findings from miRNA profiling studies |
|
 |
|
Legend:
1 Non-obese diabetic. 2 Prediabetic and diabetic. 3 Before and after 3-hr euglycemic-hyperinsulinemic clamp. 4 Skeletal muscle. 5 Hyperglycemic vs. intermediate glycemic (Wistar Kyoto) and normoglycemic (Brown Norway) rats. |
|
2.3 miRNA dysregulation in insulin-sensitive peripheral tissues
Glucose homeostasis is maintained by a balance between the amount of insulin released by pancreatic β-cells and the action of the hormone at insulin-sensitive target tissues. During the development of insulin resistance, target tissues become less sensitive to insulin, which leads to progressive hyperglycemia. Age, obesity, diet, and hypertension are known to affect insulin sensitivity of target tissues, but the precise mechanisms by which insulin resistance develops remain an area of active focus. Just as miRNAs contribute to dysregulation of β-cell function, there is now compelling evidence supporting a role for these ncRNAs in the development of insulin resistance (Table 1).
For example, expression of miR-29a/b/c was elevated in muscle, fat, and/or liver of diabetic rats [86], fa/fa rats, and high-fat diet-fed mice [87]. Overexpression of miR-29a/b/c in 3T3-L1 adipocytes decreased insulin-stimulated glucose uptake by inhibiting p85alpha and AKT activation [86, 88], and led to insulin resistance through mechanisms not involving AKT as the direct target of the miRNA [86]. Overexpression of miR-29a in HepG2 cells prevented insulin-mediated inhibition of the phosphoenolpyruvate carboxykinase gene (PEPCK) by repressing the p85alpha gene, the upstream intermediate of AKT [89]. As noted above, miR-29a/b/c also plays a known role in insulin signaling through mechanisms involving MCT1 [63, 90].
Another miRNA, miR-320, has also been shown to play a role in the regulation of insulin sensitivity. Expression of miR-320 in insulin resistant 3T3-L1 adipocytes was 50-fold higher compared to normal adipocytes [91]. Inhibition of this miRNA in 3T3-L1 cells led to improved insulin sensitivity. The p85 subunit of phosphatidylinositol 3-kinase (PI3-K) was identified as a potential mRNA target, and knockdown of miR-320 not only corresponded with increased expression of p85, but also with elevated expression of glucose transporter type 4 (GLUT-4), improved insulin-stimulated glucose uptake, and elevated phosphorylation of AKT [91]. Notably, these findings were observed only in insulin-resistant 3T3-L1 cells and not in normal adipocytes, suggesting that the dysregulation of insulin action leads to alterations in the miR-320/p85 pathway that further exacerbate the condition of insulin resistance.
Transgenic mice overexpressing lin28a displayed improved glucose tolerance and were resistant to high fat diet-induced diabetes, while animals with muscle-specific lin28a deletion and inducible let-7 exhibited glucose intolerance [92]. Functional analyses demonstrated that lin28a, which blocks let-7 miRNAs, increases glucose uptake through insulin-PI3K-mTOR signaling by depressing direct let-7 targets in this pathway, including insulin-like growth factor 1 (IGF1R), insulin receptor (INSR), insulin receptor substrate 2 (IRS2), phosphoinositide-3-kinase interacting protein 1 (PIK3IP1), v-akt murine thymoma viral oncogene homolog 2 (AKT2), tuberous sclerosis 1 (TSC1) and RPTOR independent companion of MTOR, complex 2 (RICTOR). Inhibition of mTOR demonstrated a role for Lin28a glucose tolerance and insulin sensitivity in an mTOR-dependent response [92]. These results provide strong evidence that the Lin28/let-7 pathway may be a prime factor in the regulation of glucose metabolism.
Granjon et al. compared miRNA expression profiles in skeletal muscle biopsies from healthy individuals before and after a 3-hour euglycemic-hyperinsulinemic clamp, and observed downregulation of three miRNAs by insulin: miR-1, miR-133a, and miR-306 [93]. miR-1 and miR-133a both play roles in myoblast biology, but while miR-1 inhibits myoblast proliferation, miR-133a enhances myoblast growth, resulting in differentiation and proliferation [94]. Both miR-1 and miR-133a are directly activated by myocyte enhancer factor 2C (MEF2C), which is a major regulator of muscle development [95]. The authors determined that it is through the combined repression of MEF2C and activation of sterol regulatory element binding protein 1c (SREBP1c) that insulin downregulates miR-1 and miR-133a [93]. In streptozotocin-treated mice, levels of miR-1 and miR-133a were significantly higher compared with control animals, but miR-133 expression was not altered in healthy human subjects during a hyperglycemic-euglycemic clamp, indicating that hyperglycemia, in the absence of hyperinsulinemia, does not appear to affect miR-133a levels in humans [93]. In contrast, skeletal muscle expression of miR-133a was significantly different among individuals with normal glucose tolerance, impaired glucose tolerance, and T2D, and an association between higher fasting glucose levels and lower miR-133a expression was also observed in these patients [96].
A separate study by Gallagher and colleagues reported differential expression of 62 out of 171 miRNAs in skeletal muscle from individuals with T2D [96]. Of these, ~15% of the dysregulated miRNAs showed differential expression patterns early in the disease process. Using bioinformatics approaches, the authors determined that the potential combinatorial nature of miRNA action in vivo underlies significant changes in target protein levels, which then contributes to the development of insulin resistance and T2D. In an analysis of 283 miRNAs in adipose tissue, skeletal muscle, and liver from hyperglycemic (GK), intermediate glycemic (Wistar Kyoto), and normoglycemic (Brown Norway) rats, 49 differentially expressed miRNAs were identified across the three tissues [97]. Of these, only five had expression levels that correlated with degree of glycemia, including miR-222 and miR-27a, which were upregulated in adipose tissue, miR-195 and miR-103, which were upregulated in liver, and miR-10b, which was downregulated in muscle. Expression patterns for miR-222, miR-27a, and miR-29a were validated in 3T3-L1 adipocytes cultured under hyperglycemic conditions, suggesting that altered miRNA expression may occur early in the pathogenesis of T2D.
An examination of liver-specific miRNA expression in ob/ob and diet-induced obese mice identified miR-103/miR-107 as two of the most significantly upregulated miRNAs [98], validating findings from hyperglycemic GK rats [97]. Of note, the sequences of mature miR-103 and miR-107 differ only at one nucleotide, and are thus nearly indistinguishable from each other. Expression of miR-103 and miR-107 was also increased in liver biopsies from individuals with non-alcoholic fatty liver disease, which often accompanies obesity and T2D, and levels of these miRNAs were positively correlated with homeostatic model assessment (HOMA) index [98]. Overexpression of miR-107 in the liver resulted in excess glucose output through a mechanism involving increased gluconeogenesis. Knockdown of miR-103 and miR-107 corresponded with improved insulin signaling in liver and adipose tissue, although targeted silencing of these miRNAs in the liver did not ameliorate the metabolic abnormalities observed in these animals. The investigators identified caveolin-1 as a direct target of miR-103/miR-107. Inhibition of miR-103/miR-107 in adipocytes led to upregulation of caveolin-1 expression, which was accompanied by insulin receptor stabilization, improved insulin signaling, decreased adipocyte size, and enhanced insulin-stimulated glucose uptake. Elucidation of the mechanism by which the miR-103/107-caveolin interaction regulates insulin signaling may have important implications for the development of these miRNAs as potential targets for T2D treatment [98].
A sequencing analysis of small RNA molecules extracted from the livers of mice with either free or restricted access to food, as well as fasted/refed animals, found that out of 32 confirmed miRNAs, only one, miR-143, varied with feeding conditions [99]. In db/db mice and mice fed a high-fat diet, hepatic miR-143 expression was upregulated. Another miRNA, miR-145, which is located in the same gene cluster as miR-143, was also upregulated in livers of these two animal models. Inhibition of miR-143 expression protected mice from diet-induced insulin resistance and AKT activation, while miRNA overexpression disrupted insulin-stimulated AKT activation and led to impaired glucose metabolism [99]. A potential miR-143 target, oxysterol-binding protein-related protein 8 (ORP8), was identified, and decreased hepatic expression of this target led to impaired insulin-induced AKT activation. A similar sequence analysis of hepatic ncRNA in ob/ob and control mice revealed 37 differentially expressed miRNAs [100], with miR-122 showing the greatest expression changes between the two groups, followed by miR-24, miR-195a, miR-106b, miR-15b, miR-802, miR-185, miR-214, miR-378, and let-7c (upregulated), and miR-224, miR-126, miR-7a, miR-128, miR-455, miR-452, miR-135b, miR-145, miR-18a, and miR-196a (downregulated).
2.4 Genetic variation, miRNAs, and susceptibility to T2D
Polymorphisms located in regulatory regions of miRNA genes can affect miRNA processing and expression, while those in the miRNA seed region can potentially attenuate or abolish miRNA binding to target mRNAs or create new interactions with non-target mRNAs [101, 102]. Likewise, variants in miRNA target sites may disrupt gene expression, leading to altered risk of developing disease [103-105]. Given the importance of miRNAs in the regulation of homeostatic processes, it is not surprising that miRNA-related polymorphisms have been associated with a diverse array of diseases [106-108]. Over the past two years, growing evidence is beginning to support a role for genetic variation in miRNAs and miRNA target sites, yielding an additional level of complexity associated with the development of T2D.
In their analysis of human islet and β-cell miRNAs discussed above, van de Bunt and colleagues [31] identified over 6000 variants in miRNA transcripts and predicted miRNA target sites overlapping genome-wide association data from the Diabetes Genetics Replication and Meta-analysis (DIAGRAM) consortium [109]. Of these, rs3802177, located in the 3'UTR of SLC30A8, was the only variant that reached genome-wide levels of statistical significance (p = 1.45 x 10-8). The sequence harboring this variant corresponded to predicted target sites for several miRNAs including miR-363-3p, miR-25-3p, miR-32-5p, miR-92a-3p, miR-33a-5p, and miR-33b-5p. The authors also observed that predicted mRNA targets of islet miRNAs were enriched for T2D association signals and identified overlap between variants in miRNA sequence and/or predicted miRNA targets and six loci (AP3S2, KCNK16, NOTCH2, SLC30A8, VPS26A, and WFS1) showing genome-wide evidence for association with T2D [31].
In a study of 1017 T2D cases and 1059 controls, Zhao et al. analyzed ten polymorphisms located in diabetes-related miRNA target-binding sites [110]. They found an association between variants in the insulin receptor gene (rs1366600), acyl-CoA synthetase 1 (rs2292899), and the fatty acid-binding protein 2 gene (rs11724758). These variants affect the sequences of specific miRNA-binding sites (miR-20b, miR-34a, and miR-132, respectively), but the functional consequences of these findings remain to be determined. Using a different approach, Ciccacci and colleagues [32] sequenced 13 miRNA genes and identified 15 polymorphisms, including nine known and six novel variants, although none of the markers was located within miRNA seed regions. In an analysis of the variants in 165 individuals with T2D and 185 healthy controls, the authors observed evidence for association with the G allele of rs895819 in miR-27a (OR = 0.58; p = 0.008) and the G allele of rs531564 in miR-124a (OR = 2.15; p = 0.008). While this study was the first to directly examine potential associations between miRNA polymorphisms and T2D, the sample size used was relatively small and the number of miRNAs investigated was limited. Additional studies will be necessary to confirm the major findings and a global assessment of miRNA-specific variants may likely identify novel disease associations.
In a similar study, Zhang et al. genotyped 588 T2D patients and 588 age- and sex-matched controls regarding two variants in the let-7/Lin28 pathway (rs3811463 and rs3811464) [111]. Evidence for association was observed with rs3811463 (OR = 1.47; p = 0.005), but not rs3811464, which is consistent with an earlier study showing association of the C allele of rs3811463 with insulin sensitivity and resistance to high fat diet-induced diabetes in mice [92]. Combined with the findings showing decreased susceptibility to diet-induced diabetes in mice discussed above [69], these results suggest that genetic variation in the let-7/Lin28 pathway may increase risk of T2D. Finally, in an investigation of miRNA variants on miRNA expression, Locke et al. demonstrated that the minor allele of rs72631823 was associated with higher expression of miR-34a in MIN6 cells, compared to the major allele [112]. Given the evidence that small changes in miR-34a can significantly impact on β-cell apoptosis [67, 113], it is possible that the minor allele of rs72631823 may exert strong functional consequences in islets. Further functional characterization of this locus will enhance our understanding of this variant.
3. lncRNAs and T2D pathogenesis
Although far less is known of the impact of lncRNAs in the development of T2D compared to miRNAs, there is growing evidence that these molecules also contribute to β-cell dysfunction in response to hyperglycemia. A recent genome-wide search for human pancreatic islet and β-cell lncRNAs identified 1128 intergenic and antisense islet-cell lncRNAs of which 55% and 40% were islet-specific, respectively, compared to only 9.4% of RefSeq annotated genes [114]. Interestingly, this work showed that >19% of the transcribed genome in islets mapped outside of annotated protein-coding genes. The majority of lncRNAs identified in this study were silent or expressed at low levels in pancreatic progenitors, but were active in adult islets, indicating that lncRNAs contribute in a meaningful manner to pancreatic endocrine differentiation. Similarly, during in vivo differentiation of human embryonic stem cells, six lncRNAs were expressed at very low or undetectable levels throughout all in vitro differentiation steps, and were only activated during the in vivo maturation step [114]. In a comparison of 14 islet-specific lncRNAs, KCNQ1OT1 and HI-LNC45 were significantly increased or decreased in T2D islets, respectively, and out of 55 T2D susceptibility loci, nine contained islet lncRNAs within 150 kb of the reported lead marker, six of which have been linked directly to β-cell dysfunction [109, 115-118]. Finally, as a first step toward understanding islet lncRNA function, the authors suppressed expression of a candidate lncRNA, HI-LNC25, in EndoC-βH1 cells, a human β-cell line that exhibits glucose-stimulated insulin secretion, and examined subsequent effects on 24 islet mRNAs. Levels of GLIS3, an islet transcription factor that is mutated in monogenic diabetes, were significantly reduced in response to HI-LNC25 knockdown, imparting a novel regulatory function to islet lncRNAs.
In a comprehensive DNA and RNA sequencing effort using pancreatic islets from 89 donors with different degrees of glucose tolerance, Fadista and colleagues [119] identified a number of genetic variants regulating gene expression (eQTLs) and exon use (sQTLs), including many lincRNAs. A total of 493 RefSeq islet lincRNAs were identified, 54 of which were associated with eQTLs and sQTLs. Seventeen lincRNAs were associated with HbA1c levels, including HI-LNC901 (i.e., LOC283177), which also had an eQTL (rs73036390) and whose expression was directly correlated with insulin exocytosis. HI-LNC901 was coexpressed with MAP-kinase activating death domain (MADD), synaptotagmin 11 (SYT11), and paired box 6 (PAX6), all of which have been implicated in islet function.
Other lncRNAs have been found to harbor genetic variants associated with T2D, the most notable of which is the ANRIL locus [40]. This lncRNA maps to the INK4 locus, which encodes three tumor suppressor genes including p15INK4B. ANRIL is required for the silencing of this tumor suppressor gene [120], and it is possible that variants that disrupt the expression or function of this lncRNA may affect compensatory increases in pancreatic β-cell mass in response to increasing demands for insulin in the prediabetic state [121].
The discovery of dysregulated islet-specific lncRNAs adds a new layer of complexity to the molecular etiology of T2D. The studies reported to date, although limited in number, not only point to a role for lncRNAs in the regulation of β-cell identity and function, but also suggest that variants in islet-specific lncRNAs contribute to β-cell physiology and T2D. Functional characterization of islet-specific lncRNAs is underway [114], although a substantial amount of work is needed to understand the relative importance of these molecules in the pathogenesis of T2D. These findings, in combination with emerging results, are expected to yield new insights into the complex pathogenesis of T2D and may eventually lead to the identification of novel islet-specific therapeutic targets with limited effects in other cell types.
4. Summary
The role of ncRNAs in the pathogenesis of T2D is just beginning to emerge, but there is already ample evidence of their involvement in pathophysiological mechanisms underlying the disease. A number of miRNAs have emerged as key players in the control of β-cell function and regulation of insulin sensitivity and/or action in peripheral organs, and more will undoubtedly be identified as research in this area matures. While studies from knockout animals have demonstrated clear roles for many of these miRNAs in processes contributing to T2D pathogenesis, much less is known of the specific targets of candidate molecules and the mechanisms by which they affect disease development in susceptible individuals. Studies aimed at delineating specific miRNA/mRNA networks will enhance our understanding of the complex pathogenesis of T2D and enable exploitation of relevant miRNAs as novel targets for therapeutic interventions. The investigation of polymorphisms in miRNAs and miRNA targets, which represents an increasingly active field of research, also promises to yield new insights into the complex nature of genetic predisposition to T2D. The growing recognition of lncRNA involvement in the pathogenesis of T2D adds an exciting new layer of complexity to our understanding of both the disease and gene regulation, and the cell-specific expression patterns of these molecules may yield deeper insights into defects in specialized cellular functions.
Disclosures: The author reports no conflict of interests.
References
- DeFronzo RA, Abdul-Ghani M. Type 2 diabetes can be prevented with early pharmacological intervention. Diabetes Care 2011. 34(Suppl 2):S202-S209. [DOD] [CrossRef]
- DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care 1992. 15(3):318-368. [DOD] [CrossRef]
- Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011. 378(9785):31-40. [DOD] [CrossRef]
- Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract 2011. 94(3):311-321. [DOD] [CrossRef]
- Global status report on noncommunicable diseases 2010. 2011, World Health Organization, Geneva. [DOD]
- Berends LM, Ozanne SE. Early determinants of type-2 diabetes. Best Pract Res Clin Endocrinol Metab 2012. 26(5):569-580. [DOD] [CrossRef]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007. 316(5829):1341-1345. [DOD] [CrossRef]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, et al. A genome-wide association study identified novel risk loci for type 2 diabetes. Nature 2007. 445:881-885. [DOD] [CrossRef]
- Zeggini E, Scott LJ, Saxena R, Voight BF, Diabetes Genetics Replication and Meta-analysis C. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 2008. 40(5):638-645. [DOD]
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JRB, Rayner NW, Freathy RM, et al. Replication of genome-wide association signals in U.K. samples reveals risk loci for type 2 diabetes. Science 2007. 316(5829):1336-1341. [DOD] [CrossRef]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012. 489(7414):57-74. [DOD] [CrossRef]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009. 106(23):9362-9367. [DOD] [CrossRef]
- Maher B. ENCODE: The human encyclopaedia. Nature 2012. 489(7414):46-48. [DOD] [CrossRef]
- Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 2009. 58(12):2718-2725. [DOD] [CrossRef]
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet 2011. 12(12):861-874. [DOD] [CrossRef]
- Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet 2006. 15(Spec No 1):R17-R29. [DOD] [CrossRef]
- Kim DH, Saetrom P, Snove O Jr, Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci U S A 2008. 105(42):16230-16235. [DOD] [CrossRef]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009. 136(2):215-233. [DOD] [CrossRef]
- Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 2011. 12(2):99-110. [DOD] [CrossRef]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet 2005. 37(5):495-500. [DOD] [CrossRef]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005. 120(1):15-20. [DOD] [CrossRef]
- Natarajan R, Putta S, Kato M. MicroRNAs and diabetic complications. J Cardiovasc Transl Res 2012. 5(4):413-422. [DOD] [CrossRef]
- Tang X, Tang G, Ozcan S. Role of microRNAs in diabetes. Biochim Biophys Acta 2008. 1779(11):697-701. [DOD] [CrossRef]
- Kalis M, Bolmeson C, Esguerra JL, Gupta S, Edlund A, Tormo-Badia N, Speidel D, Holmberg D, Mayans S, Khoo NK, et al. Beta-cell specific deletion of Dicer1 leads to defective insulin secretion and diabetes mellitus. Plos One 2011. 6(12):e29166. [DOD] [CrossRef]
- Lynn FC, Skewes-Cox P, Kosaka Y, McManus MT, Harfe BD, German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes 2007. 56(12):2938-2945. [DOD] [CrossRef]
- Melkman-Zehavi T, Oren R, Kredo-Russo S, Shapira T, Mandelbaum AD, Rivkin N, Nir T, Lennox KA, Behlke MA, Dor Y, Hornstein E. miRNAs control insulin content in pancreatic beta-cells via downregulation of transcriptional repressors. EMBO J 2011. 30(5):835-845. [DOD] [CrossRef]
- Kaspi H, Pasvolsky R, Hornstein E. Could microRNAs contribute to the maintenance of beta cell identity? Trends Endocrinol Metab 2014. 25(6):285-292. [DOD]
- Guay C, Jacovetti C, Nesca V, Motterle A, Tugay K, Regazzi R. Emerging roles of non-coding RNAs in pancreatic beta-cell function and dysfunction. Diabetes Obes Metab 2012. 14(Suppl 3):12-21. [DOD] [CrossRef]
- Park SY, Jeong HJ, Yang WM, Lee W. Implications of microRNAs in the pathogenesis of diabetes. Arch Pharm Res 2013. 36(2):154-166. [DOD] [CrossRef]
- Rome S. Are extracellular microRNAs involved in type 2 diabetes and related pathologies? Clin Biochem 2013. 46(10-11):937-945. [DOD]
- van de Bunt M, Gaulton KJ, Parts L, Moran I, Johnson PR, Lindgren CM, Ferrer J, Gloyn AL, McCarthy MI. The miRNA profile of human pancreatic islets and beta-cells and relationship to type 2 diabetes pathogenesis. Plos One 2013. 8(1):e55272. [DOD] [CrossRef]
- Ciccacci C, Di Fusco D, Cacciotti L, Morganti R, D'Amato C, Greco C, Rufini S, Novelli G, Sangiuolo F, Spallone V, Borgiani P. MicroRNA genetic variations: association with type 2 diabetes. Acta Diabetol 2013. 50(6):867-872. [DOD] [CrossRef]
- Louro R, Smirnova AS, Verjovski-Almeida S. Long intronic noncoding RNA transcription: expression noise or expression choice? Genomics 2009. 93(4):291-298. [DOD]
- Moran VA, Perera RJ, Khalil AM. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res 2012. 40(14):6391-6400. [DOD] [CrossRef]
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet 2009. 10(3):155-159. [DOD] [CrossRef]
- Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell 2009. 136(4):629-641. [DOD] [CrossRef]
- Yan B, Wang Z. Long noncoding RNA: its physiological and pathological roles. DNA Cell Biol 2012. 31(Suppl 1):S34-S41. [DOD]
- Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell 2011. 145(2):178-181. [DOD] [CrossRef]
- Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell 2011. 43(6):904-914. [DOD] [CrossRef]
- Pasmant E, Sabbagh A, Vidaud M, Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J 2011. 25(2):444-448. [DOD] [CrossRef]
- Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol 2011. 21(6):354-361. [DOD] [CrossRef]
- Dumortier O, Van Obberghen E. MicroRNAs in pancreas development. Diabetes Obes Metab 2012. 14(Suppl 3):22-28. [DOD] [CrossRef]
- Eliasson L, Esguerra JL. Role of non-coding RNAs in pancreatic beta-cell development and physiology. Acta Physiol (Oxf) 2014. 211(2):273-284. [DOD] [CrossRef]
- Plaisance V, Waeber G, Regazzi R, Abderrahmani A. Role of microRNAs in islet beta-cell compensation and failure during diabetes. J Diabetes Res 2014. 2014:618652. [DOD]
- Matsuda S, Ichigotani Y, Okuda T, Irimura T, Nakatsugawa S, Hamaguchi M. Molecular cloning and characterization of a novel human gene (HERNA) which encodes a putative RNA-helicase. Biochim Biophys Acta 2000. 1490(1-2):163-169. [DOD] [CrossRef]
- Yan Y, Salazar TE, Dominguez JM 2nd, Nguyen DV, Li Calzi S, Bhatwadekar AD, Qi X, Busik JV, Boulton ME, Grant MB. Dicer expression exhibits a tissue-specific diurnal pattern that is lost during aging and in diabetes. Plos One 2013. 8(11):e80029. [DOD] [CrossRef]
- Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004. 432(7014):226-230. [DOD] [CrossRef]
- El Ouaamari A, Baroukh N, Martens GA, Lebrun P, Pipeleers D, van Obberghen E. miR-375 targets 3'-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes 2008. 57(10):2708-2717. [DOD] [CrossRef]
- Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, Zavolan M, Stoffel M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci U S A 2009. 106(14):5813-5818. [DOD] [CrossRef]
- Kloosterman WP, Lagendijk AK, Ketting RF, Moulton JD, Plasterk RH. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. Plos Biol 2007. 5(8):e203. [DOD] [CrossRef]
- Chang X, Li S, Li J, Yin L, Zhou T, Zhang C, Chen X, Sun K. Ethnic differences in microRNA-375 expression level and DNA methylation status in type 2 diabetes of Han and Kazak populations. J Diabetes Res 2014. 2014:761938. [DOD] [CrossRef]
- Sun K, Chang X, Yin L, Li J, Zhou T, Zhang C, Chen X. Expression and DNA methylation status of microRNA-375 in patients with type 2 diabetes mellitus. Mol Med Rep 2014. 9(3):967-972. [DOD]
- Zhao H, Guan J, Lee HM, Sui Y, He L, Siu JJ, Tse PP, Tong PC, Lai FM, Chan JC. Up-regulated pancreatic tissue microRNA-375 associates with human type 2 diabetes through beta-cell deficit and islet amyloid deposition. Pancreas 2010. 39(6):843-846. [DOD] [CrossRef]
- Sebastiani G, Po A, Miele E, Ventriglia G, Ceccarelli E, Bugliani M, Marselli L, Marchetti P, Gulino A, Ferretti E, Dotta F. MicroRNA-124a is hyperexpressed in type 2 diabetic human pancreatic islets and negatively regulates insulin secretion. Acta Diabetol 2014. In press. [DOD]
- Wang X, Chang X, Li J, Yin L, Sun K. DNA methylation of in impaired glucose tolerance. Exp Ther Med 2014. 8(3):775-780. [DOD]
- Li X. MiR-375, a microRNA related to diabetes. Gene 2014. 533(1):1-4. [DOD] [CrossRef]
- Latreille M, Hausser J, Stutzer I, Zhang Q, Hastoy B, Gargani S, Kerr-Conte J, Pattou F, Zavolan M, Esguerra JL, et al. MicroRNA-7a regulates pancreatic beta cell function. J Clin Invest 2014. 124(6):2722-2735. [DOD] [CrossRef]
- Baroukh N, Ravier MA, Loder MK, Hill EV, Bounacer A, Scharfmann R, Rutter GA, Van Obberghen E. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem 2007. 282(27):19575-19588. [DOD] [CrossRef]
- Jing G, Westwell-Roper C, Chen J, Xu G, Verchere CB, Shalev A. Thioredoxin-interacting protein promotes islet amyloid polypeptide expression through miR-124a and FoxA2. J Biol Chem 2014. 289(17):11807-11815. [DOD] [CrossRef]
- Bolmeson C, Esguerra JL, Salehi A, Speidel D, Eliasson L, Cilio CM. Differences in islet-enriched miRNAs in healthy and glucose intolerant human subjects. Biochem Biophys Res Commun 2011. 404(1):16-22. [DOD] [CrossRef]
- Morita S, Horii T, Kimura M, Hatada I. MiR-184 regulates insulin secretion through repression of Slc25a22. PeerJ 2013. 1:e162. [DOD] [CrossRef]
- Nesca V, Guay C, Jacovetti C, Menoud V, Peyot ML, Laybutt DR, Prentki M, Regazzi R. Identification of particular groups of microRNAs that positively or negatively impact on beta cell function in obese models of type 2 diabetes. Diabetologia 2013. 56(10):2203-2212. [DOD] [CrossRef]
- Pullen TJ, da Silva Xavier G, Kelsey G, Rutter GA. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol Cell Biol 2011. 31(15):3182-3194. [DOD] [CrossRef]
- Zhu Y, You W, Wang H, Li Y, Qiao N, Shi Y, Zhang C, Bleich D, Han X. MicroRNA-24/MODY gene regulatory pathway mediates pancreatic beta-cell dysfunction. Diabetes 2013. 62(9):3194-3206. [DOD] [CrossRef]
- Locke JM, da Silva Xavier G, Dawe HR, Rutter GA, Harries LW. Increased expression of miR-187 in human islets from individuals with type 2 diabetes is associated with reduced glucose-stimulated insulin secretion. Diabetologia 2014. 57(1):122-128. [DOD] [CrossRef]
- Shojima N, Hara K, Fujita H, Horikoshi M, Takahashi N, Takamoto I, Ohsugi M, Aburatani H, Noda M, Kubota N, et al. Depletion of homeodomain-interacting protein kinase 3 impairs insulin secretion and glucose tolerance in mice. Diabetologia 2012. 55(12):3318-3330. [DOD] [CrossRef]
- Roggli E, Britan A, Gattesco S, Lin-Marq N, Abderrahmani A, Meda P, Regazzi R. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic beta-cells. Diabetes 2010. 59(4):978-986. [DOD] [CrossRef]
- Bravo-Egana V, Rosero S, Klein D, Jiang Z, Vargas N, Tsinoremas N, Doni M, Podetta M, Ricordi C, Molano RD, et al. Inflammation-Mediated Regulation of MicroRNA Expression in Transplanted Pancreatic Islets. J Transplant 2012. 2012:723614. [DOD] [CrossRef]
- Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci U S A 2011. 108(52):21075-21080. [DOD] [CrossRef]
- Klein D, Misawa R, Bravo-Egana V, Vargas N, Rosero S, Piroso J, Ichii H, Umland O, Zhijie J, Tsinoremas N, et al. MicroRNA expression in alpha and beta cells of human pancreatic islets. Plos One 2013. 8(1):e55064. [DOD] [CrossRef]
- Esguerra JL, Bolmeson C, Cilio CM, Eliasson L. Differential glucose-regulation of microRNAs in pancreatic islets of non-obese type 2 diabetes model Goto-Kakizaki rat. Plos One 2011. 6(4):e18613. [DOD] [CrossRef]
- Tang X, Muniappan L, Tang G, Ozcan S. Identification of glucose-regulated miRNAs from pancreatic beta cells reveals a role for miR-30d in insulin transcription. RNA 2009. 15(2):287-293. [DOD] [CrossRef]
- Zhao E, Keller MP, Rabaglia ME, Oler AT, Stapleton DS, Schueler KL, Neto EC, Moon JY, Wang P, Wang IM, et al. Obesity and genetics regulate microRNAs in islets, liver, and adipose of diabetic mice. Mamm Genome 2009. 20(8):476-485. [DOD] [CrossRef]
- Kameswaran V, Bramswig NC, McKenna LB, Penn M, Schug J, Hand NJ, Chen Y, Choi I, Vourekas A, Won KJ, et al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab 2014. 19(1):135-145. [DOD] [CrossRef]
- Dorrell C, Abraham SL, Lanxon-Cookson KM, Canaday PS, Streeter PR, Grompe M. Isolation of major pancreatic cell types and long-term culture-initiating cells using novel human surface markers. Stem Cell Res 2008. 1(3):183-194. [DOD] [CrossRef]
- Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, Guo J, Zhang Y, Chen J, Guo X, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res 2008. 18(10):997-1006. [DOD] [CrossRef]
- Corral-Fernandez NE, Salgado-Bustamante M, Martinez-Leija ME, Cortez-Espinosa N, Garcia-Hernandez MH, Reynaga-Hernandez E, Quezada-Calvillo R, Portales-Perez DP. Dysregulated miR-155 expression in peripheral blood mononuclear cells from patients with type 2 diabetes. Exp Clin Endocrinol Diabetes 2013. 121(6):347-353. [DOD] [CrossRef]
- Karolina DS, Armugam A, Tavintharan S, Wong MT, Lim SC, Sum CF, Jeyaseelan K. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. Plos One 2011. 6(8):e22839. [DOD] [CrossRef]
- Kong L, Zhu J, Han W, Jiang X, Xu M, Zhao Y, Dong Q, Pang Z, Guan Q, Gao L, et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol 2011. 48(1):61-69. [DOD] [CrossRef]
- Ortega FJ, Mercader JM, Moreno-Navarrete JM, Rovira O, Guerra E, Esteve E, Xifra G, Martinez C, Ricart W, Rieusset J, et al. Profiling of circulating microRNAs reveals common microRNAs linked to type 2 diabetes that change with insulin sensitization. Diabetes Care 2014. 37(5):1375-1383. [DOD] [CrossRef]
- Pescador N, Perez-Barba M, Ibarra JM, Corbaton A, Martinez-Larrad MT, Serrano-Rios M. Serum circulating microRNA profiling for identification of potential type 2 diabetes and obesity biomarkers. Plos One 2013. 8(10):e77251. [DOD] [CrossRef]
- Rong Y, Bao W, Shan Z, Liu J, Yu X, Xia S, Gao H, Wang X, Yao P, Hu FB, Liu L. Increased microRNA-146a levels in plasma of patients with newly diagnosed type 2 diabetes mellitus. Plos One 2013. 8(9):e73272. [DOD] [CrossRef]
- Wang X, Sundquist J, Zoller B, Memon AA, Palmer K, Sundquist K, Bennet L. Determination of 14 circulating microRNAs in Swedes and Iraqis with and without diabetes mellitus type 2. Plos One 2014. 9(1):e86792. [DOD] [CrossRef]
- Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, Mayr A, Weger S, Oberhollenzer F, Bonora E, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res 2010. 107(6):810-817. [DOD] [CrossRef]
- Zhang T, Lv C, Li L, Chen S, Liu S, Wang C, Su B. Plasma miR-126 is a potential biomarker for early prediction of type 2 diabetes mellitus in susceptible individuals. Biomed Res Int 2013. 2013:761617. [DOD]
- He A, Zhu L, Gupta N, Chang Y, Fang F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol Endocrinol 2007. 21(11):2785-2794. [DOD] [CrossRef]
- Kurtz CL, Peck BC, Fannin EE, Beysen C, Miao J, Landstreet SR, Ding S, Turaga V, Lund PK, Turner S, et al. MicroRNA-29 fine-tunes the expression of key FOXA2-activated lipid metabolism genes and is dysregulated in animal models of insulin resistance and diabetes. Diabetes 2014. 63(9):3141-3148. [DOD] [CrossRef]
- Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol 2009. 16(1):23-29. [DOD] [CrossRef]
- Pandey AK, Verma G, Vig S, Srivastava S, Srivastava AK, Datta M. miR-29a levels are elevated in the db/db mice liver and its overexpression leads to attenuation of insulin action on PEPCK gene expression in HepG2 cells. Mol Cell Endocrinol 2011. 332(1-2):125-133. [DOD] [CrossRef]
- Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, Kere J, Sipila I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 2003. 52(1):199-204. [DOD] [CrossRef]
- Ling HY, Ou HS, Feng SD, Zhang XY, Tuo QH, Chen LX, Zhu BY, Gao ZP, Tang CK, Yin WD, et al. CHANGES IN microRNA (miR) profile and effects of miR-320 in insulin-resistant 3T3-L1 adipocytes. Clin Exp Pharmacol Physiol 2009. 36(9):e32-e39. [DOD] [CrossRef]
- Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011. 147(1):81-94. [DOD] [CrossRef]
- Granjon A, Gustin MP, Rieusset J, Lefai E, Meugnier E, Guller I, Cerutti C, Paultre C, Disse E, Rabasa-Lhoret R, et al. The microRNA signature in response to insulin reveals its implication in the transcriptional action of insulin in human skeletal muscle and the role of a sterol regulatory element-binding protein-1c/myocyte enhancer factor 2C pathway. Diabetes 2009. 58(11):2555-2564. [DOD] [CrossRef]
- Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 2006. 38(2):228-233. [DOD] [CrossRef]
- Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, Richardson JA, Bassel-Duby R, Olson EN. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc Natl Acad Sci U S A 2007. 104(52):20844-20849. [DOD] [CrossRef]
- Gallagher IJ, Scheele C, Keller P, Nielsen AR, Remenyi J, Fischer CP, Roder K, Babraj J, Wahlestedt C, Hutvagner G, et al. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Med 2010. 2(2):9. [DOD] [CrossRef]
- Herrera BM, Lockstone HE, Taylor JM, Ria M, Barrett A, Collins S, Kaisaki P, Argoud K, Fernandez C, Travers ME, et al. Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia 2010. 53(6):1099-1109. [DOD] [CrossRef]
- Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH, Stoffel M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011. 474(7353):649-653. [DOD] [CrossRef]
- Jordan SD, Kruger M, Willmes DM, Redemann N, Wunderlich FT, Bronneke HS, Merkwirth C, Kashkar H, Olkkonen VM, Bottger T, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol 2011. 13(4):434-446. [DOD] [CrossRef]
- Liang T, Liu C, Ye Z. Deep sequencing of small RNA repertoires in mice reveals metabolic disorders-associated hepatic miRNAs. Plos One 2013. 8(11):e80774. [DOD] [CrossRef]
- Chen K, Song F, Calin GA, Wei Q, Hao X, Zhang W. Polymorphisms in microRNA targets: a gold mine for molecular epidemiology. Carcinogenesis 2008. 29(7):1306-1311. [DOD] [CrossRef]
- Slaby O, Bienertova-Vasku J, Svoboda M, Vyzula R. Genetic polymorphisms and microRNAs: new direction in molecular epidemiology of solid cancer. J Cell Mol Med 2012. 16(1):8-21. [DOD] [CrossRef]
- Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, et al. Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science 2005. 310(5746):317-320. [DOD] [CrossRef]
- Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, Muller RU, Straka E, Su L, Burki EA, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3' untranslated region increases non-small cell lung cancer risk. Cancer Res 2008. 68(20):8535-8540. [DOD] [CrossRef]
- Mishra PJ, Humeniuk R, Mishra PJ, Longo-Sorbello GS, Banerjee D, Bertino JR. A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc Natl Acad Sci U S A 2007. 104(33):13513-13518. [DOD] [CrossRef]
- Chen AX, Yu KD, Fan L, Li JY, Yang C, Huang AJ, Shao ZM. Germline genetic variants disturbing the Let-7/LIN28 double-negative feedback loop alter breast cancer susceptibility. Plos Genet 2011. 7(9):e1002259. [DOD] [CrossRef]
- Hu Z, Chen J, Tian T, Zhou X, Gu H, Xu L, Zeng Y, Miao R, Jin G, Ma H, et al. Genetic variants of miRNA sequences and non-small cell lung cancer survival. J Clin Invest 2008. 118(7):2600-2608. [DOD]
- Yue C, Wang M, Ding B, Wang W, Fu S, Zhou D, Zhang Z, Han S. Polymorphism of the pre-miR-146a is associated with risk of cervical cancer in a Chinese population. Gynecol Oncol 2011. 122(1):33-37. [DOD] [CrossRef]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010. 42(7):579-589. [DOD] [CrossRef]
- Zhao X, Ye Q, Xu K, Cheng J, Gao Y, Li Q, Du J, Shi H, Zhou L. Single-nucleotide polymorphisms inside microRNA target sites influence the susceptibility to type 2 diabetes. J Hum Genet 2013. 58(3):135-141. [DOD] [CrossRef]
- Zhang J, Zhang L, Fan R, Guo N, Xiong C, Wang L, Jin S, Li W, Lu J. The polymorphism in the let-7 targeted region of the Lin28 gene is associated with increased risk of type 2 diabetes mellitus. Mol Cell Endocrinol 2013. 375(1-2):53-57. [DOD] [CrossRef]
- Locke JM, Lango Allen H, Harries LW. A rare SNP in pre-miR-34a is associated with increased levels of miR-34a in pancreatic beta cells. Acta Diabetol 2014. 51(2):325-329. [DOD] [CrossRef]
- Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang JY, Widmann C, Abderrahmani A, Regazzi R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008. 57(10):2728-2736. [DOD] [CrossRef]
- Moran I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T, Arnes L, Nakic N, Garcia-Hurtado J, Rodriguez-Segui S, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab 2012. 16(4):435-448. [DOD] [CrossRef]
- Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 2012. 44(1):67-72. [DOD] [CrossRef]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010. 42(2):105-116. [DOD] [CrossRef]
- Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, Been LF, Chia KS, Dimas AS, Hassanali N, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet 2011. 43(10):984-989. [DOD] [CrossRef]
- Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E, Rybin D, Petrie JR, Travers ME, Bouatia-Naji N, Dimas AS, et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011. 60(10):2624-2634. [DOD] [CrossRef]
- Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J, Storm P, Osmark P, Ladenvall C, Prasad RB, et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci U S A 2014. 111(38):13924-13929. [DOD] [CrossRef]
- Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011. 30(16):1956-1962. [DOD] [CrossRef]
- Pullen TJ, Rutter GA. Could lncRNAs contribute to beta-cell identity and its loss in Type 2 diabetes? Biochem Soc Trans 2013. 41(3):797-801. [DOD]
- Chen L, Hou J, Ye L, Chen Y, Cui J, Tian W, Li C, Liu L. MicroRNA-143 regulates adipogenesis by modulating the MAP2K5-ERK5 signaling. Sci Rep 2014. 4:3819. [DOD]
|


)
)
)

