Original Data
| Rev Diabet Stud,
2005,
2(3):146-156 |
DOI 10.1900/RDS.2005.2.146 |
Insulinotropic and Anti-Inflammatory Effects of Rosiglitazone in Experimental Autoimmune Diabetes
Wageh M. Awara1, Alaa E. El-Sisi1, Mohamed El-Refaei2, Mona M. El-Naa1, Karima El-Desoky3
1Department of Pharmacology/Toxicology, College of Pharmacy, University of Tanta, Egypt.
2Department of Biochemistry, Genetic Engineering and Biotechnology Research Institute, Minofia University, Egypt.
3Department of Pathology, Faculty of Medicine, University of Tanta, Egypt.
Address correspondence to: Wageh M. Awara, e-mail: wagawara@yahoo.com.
Keywords: autoimmune diabetes, thiazolidinedione, rosiglitazone, insulitis, PPAR-gamma, cytokines, nitric oxide, multiple low doses of streptozotocin
Abstract
Cytokines and nitric oxide (NO) are involved in the pathogenesis of autoimmune diabetes mellitus (DM). Rosiglitazone is an insulin-sensitizing drug that is a ligand for the nuclear receptor peroxisome proliferator-activated receptor-gamma (PPAR-γ). The anti-inflammatory and immunomodulating properties of PPAR-γ have been documented. The aim of this study is to investigate the effectiveness of rosiglitazone in autoimmune DM and to clarify the possible mechanism(s) involved. Autoimmune DM was induced in adult male Balb/c mice by co-administration of cyclosporin A and multiple low doses of streptozotocin. Diabetic mice were treated daily with rosiglitazone (7 mg/kg, p.o.) for 21 days. Blood glucose level (BGL), serum insulin level and pancreatic levels of tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ) and NO were measured. Histopathological examination and immunohistochemical determination of CD4 and CD8 T lymphocytes in the pancreatic islets were performed. In addition, analysis of pancreatic protein expression was carried out. The results showed that rosiglitazone treatment resulted in a significant decrease in the BGL and the pancreatic levels of TNF-α, IFN-γ and NO compared to diabetic mice. The serum insulin level was significantly increased after rosiglitazone treatment compared to diabetic mice. The destroyed pancreatic islets were regenerated and became free from both CD4 and CD8 T cells after treatment. Furthermore, many changes in pancreatic protein expression were observed. These results suggest that rosiglitazone has a beneficial effect in the treatment of autoimmune diabetes, an effect that seemed to be a secondary consequence of its anti-inflammatory and immunomodulating properties and might be reflected at the level of protein expression.
Introduction
Diabetes mellitus (DM) of type 1 is an autoimmune disease caused by selective destruction of pancreatic β-cells [1]. Pathological features of autoimmune type 1 DM include the infiltration of inflammatory cells into the islets, insulitis followed by selective destruction of β-cells [2]. Inflammatory cytokines, such as IL-1β, TNF-α and IFN-γ secreted by infiltrating immune cells, as well as free radicals, such as nitric oxide (NO), are involved in β-cell dysfunction and damage in autoimmune diabetes [2-15].
Inflammatory cytokines induce and accelerate β-cell destruction through direct cytotoxic effects via mechanisms that involve induction of free radicals and apoptosis-activating pathways in β-cells [4]. However, indirect β-cell destruction may be mediated through mech-anisms including activation of autoreactive T cells [5], suppression of the production of soluble cytokine antagonists [6] and upregulation of MHC class I and Fas receptor expression on β-cells [7].
Several experimental models that share immunopathological similarities with human type 1 DM, such as NOD mouse, diabetes-prone BB rat and multiple low doses of streptozotocin (MLDSTZ), have extensively been used as in vivo tools for gaining insights into the pathological mechanisms and for testing possible immunomodulatory compounds endowed with antidiabetogenic properties which are worthy of being considered for translation into the clinical setting [8-10].
In particular, repeated injections of MLDSTZ given to susceptible strains of mice provoke a condition with clinical, histological and immunopathogenic characteristics resembling human type 1 DM, including the development of hyperglycemia associated with infiltration of the pancreatic islets by T lymphocytes and macrophages [8, 9]. As in the NOD mouse, in the diabetes-prone BB rat, and probably in humans, the immuno-inflammatory diabetogenic process triggered by MLDSTZ appears to be related to preferential production of inflammatory cytokines and NO by islet-infiltrating mononuclear cells [11-16]. While MLDSTZ in susceptible strains of mice produced insulitis and autoimmune diabetes that required several weeks to fully develop [8, 9], resistant strains of mice became susceptible, which resulted in full-blown insulitis and diabetes caused by selective immunosuppressive drugs directed at suppressor T cells, such as cyclosporin A (CsA) [17, 18].
Thiazolidinediones (TZDs) such as rosiglitazone are new oral antidiabetic agents that have been approved for treatment of type 2 DM [19, 20]. The antidiabetic activity of TZDs is mediated through activation of the peroxisome proliferator-activated receptor-gamma (PPAR-γ) with subsequent improvement of insulin sensitivity [19, 20]. PPAR-γ is a member of the nuclear receptor superfamily of ligand-dependent transcription factors [19-21]. In addition to its involvement in glucose homeostasis, PPAR-γ is reportedly involved in lipid and lipoprotein metabolism, cell proliferation and differentiation and apoptosis [21]. Moreover, a growing body of evidence suggests that PPAR-γ may play a role in the control of inflammation [21, 22] suggesting the potential use of TZDs as anti-inflammatory drugs [22-24].
Therefore, considering the autoimmune nature of type 1 DM as well as the immunomodulatory and the anti-inflammatory properties of TZDs, this study was designed to find out the role that may be played by certain inflammatory mediators such as cytokines and NO in the development of autoimmune diabetes in mice. The role of rosiglitazone in influencing the immune system and subsequently its effectiveness in autoimmune diabetes mellitus were also assessed.
Materials and methods
Animals
Adult male Balb/C mice weighing 25-30 g (aged 6 wk) were purchased from the National Institute of Ophthalmology, Giza, Egypt. The animals were maintained under standard laboratory conditions with free access to food (standard laboratory animal chow, El-Nasr Pharmaceutical Chemicals Co., Cairo, Egypt), and water. Procedures involving animals and their care were in conformity with the institutional guidelines and in compliance with national and international laws on the care and use of laboratory animals.
Chemicals and drugs
Cyclosporin A (CsA), Sandimmune® injection, was kindly donated by Sandoz, Switzerland. Rosiglitazone was purchased from Cayman Chemical, Ann Arbor, MI, USA. TNF-α and IFN-γ detection kits were obtained from Biosource, Belgium. CD4 and CD8 T-lymphocytes detection kits were obtained from Novocastra Laboratories Ltd, UK. Insulin kits were purchased from Abbott Laboratories, USA. All the other chemicals used were purchased from Sigma Co., USA.
Induction of autoimmune type 1 DM
Thirty mice were weighed and ear-notched. STZ was dissolved in 0.1M sodium citrate buffer (pH 4.5) and injected within 15 min of preparation. Mice were administered CsA (22 mg/kg/day, s.c.) daily for 2 wk prior to STZ treatment and simultaneously MLDSTZ (45 mg/kg/day, i.p.) for 5 consecutive days [18]. Non-fasting blood samples were collected every other day via tail bleeding into heparinized tubes. Glucose concentrations in plasma samples were determined by the enzymatic colorimetric method [25]. Mice were defined as diabetic when their non-fasting blood glucose level (BGL) reached more than 200 mg/dl in two consecutive readings.
Study design
After 14 days of the last dose of CsA/MLDSTZ co-administration, mice that showed diabetes were selected (n = 26). Diabetic mice were divided into two equal groups (n = 13 for each group) and treated with either normal saline (group 1, diabetic saline) or 7 mg/kg rosiglitazone (group 2, diabetic rosiglitazone).
At the same time, twenty normal mice were divided into two equal groups (n = 10 for each group) and treated with either saline (group 3, control saline) or 7 mg/kg rosiglitazone (group 4, control rosiglitazone).
All treatments were given orally and daily for a period of 21 days. At the end of the treatment period, mice were anesthetized with ether, blood was withdrawn by heart puncture and centrifuged at 3000 rpm for 5 min. Serum was separated for determination of serum insulin levels. The pancreas from each mouse was removed and bisected longitudinally. One half was dried carefully with filter paper and homogenized in PBS to prepare a 10% homogenate. The supernatant was removed for determination of TNF-α, IFN-γ and NO levels as well as for determination of the pattern of total pancreatic protein expression by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The remaining half of each excised pancreas was immersion-fixed in 10% neutral formalin for 24 h. Sections were embedded in paraffin wax, serially sectioned (5 μm), stained with hematoxylin and eosin (H&E) for histopathological examination. In addition, immunohistochemical determination of CD4 and CD8 in the pancreatic tissues was carried out.
Determination of serum insulin levels
The insulin level in serum samples was determined using the microparticle enzyme immunoassay (MEIA), Abbott AxSYM® system.
Determination of the TNF-α and IFN-γ content in pancreatic tissue
The pancreatic levels of TNF-α and IFN-γ were determined by means of ELISA kits using monoclonal antibodies specific for TNF-α and IFN-γ respectively. Samples were run in duplicate according to the manufacturer's instructions. Concentrations of cytokines were determined from standard curves using purified recombinant cytokines provided with the kits.
Determination of the nitric oxide content in pancreatic tissue
The levels of nitric oxide in the pancreatic tissues were measured as nitrite and nitrate according to the method of Miranda et al. (2001), which is based on the Griess reaction with a prior reduction step using vanadium chloride to convert nitrates to nitrite ions [26]. The absorbance of samples was measured at 540 nm by double-beam spectrophotometer (Shimadzu UV-PC 1601, Japan).
Determination of the total protein pattern in pancreatic tissue
Proteins were separated according to their molecular weights by SDS-PAGE [27]. The molecular weight of the protein that resolved on polyacrylamide gel was determined using a gel documentation system (Amersham, Pharmacia Biotech, USA).
Histopathological examination of the pancreas
For evaluation of insulitis, at least 10 islets were examined for each treatment group. Insulitis was graded as follows [17]: negative (-) - no mononuclear cell infiltration; mild (+) - a few mononuclear cell infiltrations in and around the islet; moderate (++) - obvious cell infiltrations in and around the islet; severe (+++) - massive cell infiltrations were observed as the structure of the islet was destroyed. Pancreases were graded for insulitis on the basis of the most severely involved islet in each pancreas.
Immunohistochemical determination of CD4 and CD8 T lymphocytes infiltrating the pancreatic islets
Pancreatic tissue infiltration by CD4 and CD8 T lymphocytes was detected by immunostaining of sections prepared from formalin-fixed, paraffin-embedded pancreas based on the high-temperature antigen-unmasking technique using CD4 and CD8 detection kits, respectively, according to the manufacturer’s instructions. Grading of CD4 or CD8 lymphocytic infiltrates as either positive or negative was performed on the most heavily involved islet in each section.
Statistical analysis
Data are expressed as means ± standard error of mean (SEM). Comparisons between different groups were carried out by one-way analysis of variance (ANOVA) followed by the Tukey-Kramer test. Comparison within the same group was done by the paired Student's t-test. The level of significance was set at p < 0.05. Graphpad software instat (version 2) was used to carry out statistical analysis.
Results
Autoimmune type 1 DM model
Co-treatment of mice with CsA/MLDSTZ resulted in autoimmune type 1 DM. The suitability of this model is confirmed and validated in our laboratory biochemically by the time-dependent increase in the blood glucose level in 86.7% of mice (26 out of 30 mice) and the reduction in the serum insulin level. The elevated BGL was reduced after injection of 4 units of exogenous human insulin. In addition, histopathological examination of pancreatic sections from diabetic mice revealed early vascular degeneration with mild hyalinization of pancreatic islets of Langerhans and moderate (++) mononuclear cell infiltration (data not shown).
Effect of rosiglitazone on blood glucose level
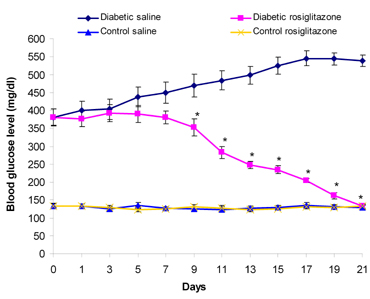
Treatment of diabetic mice with rosiglitazone (7 mg/kg/day) showed a time-dependent decrease in BGL. The reduction in BGL started to be significant by day 9 of treatment compared to the level before treatment. By day 21, the BGL was reduced to 132 ± 6.87 mg/dl, which was non-significantly different from the value in the control saline group (p > 0.05, ANOVA) (Figure 1).
 |
 |
Figure 1. Time-course effect of rosiglitazone on BGL. Mice were treated with rosiglitazone (7 mg/kg/day) for 21 days. BGL was determined every other day. Data are presented as mean ± SEM, n = 13 for diabetic groups and n = 10 for control groups. Day 0: immediately before the start of treatment. Day 1: the first day of rosiglitazone treatment. Asterisk (*) indicates significant difference from the same group before treatment (day 0). p < 0.05; paired Student's t-test. |
|
On the other hand, the BGL of diabetic mice that received saline was time-dependently increased throughout the treatment period. Normal mice treated with rosiglitazone (7 mg/kg/day) for 21 days showed non-significant changes in BGL compared to the control saline group (p > 0.05, ANOVA) (Figure 1).
Effect of rosiglitazone on serum insulin level
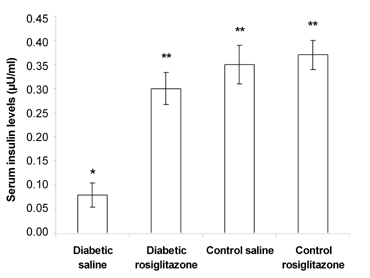
Diabetic mice that received saline for 21 days showed a significant decrease (77.1%) in the serum insulin level compared to the control saline group. On the other hand, treatment of diabetic mice with rosiglitazone (7 mg/kg/day) for 21 days resulted in a significant increase (275%) in the serum insulin level compared to the diabetic saline group (Figure 2). Normal mice treated with rosiglitazone showed a non-significant change in the serum insulin level compared to the control saline group (Figure 2).
 |
|
Figure 2. Effect of rosiglitazone on the serum insulin level in diabetic mice. Mice were treated with rosiglitazone (7 mg/kg/day) for 21 days. Serum insulin levels were determined at the end of the treatment period. Data are presented as mean ± SEM, n = 13 for diabetic groups and n = 10 for control groups. (*) indicates significant difference from control saline group. (**) indicates significant difference from diabetic saline group. p < 0.05; ANOVA, Tukey-Kramer test. |
|
Effect of rosiglitazone on the TNF-α and IFN-γ content in pancreatic tissue
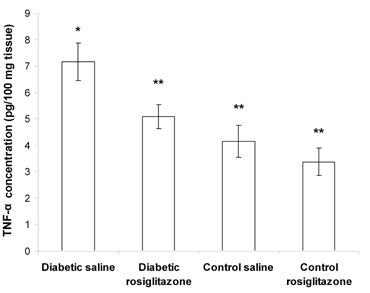
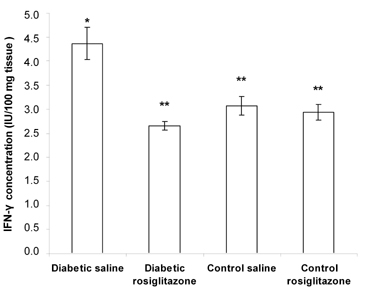
Diabetic mice that received saline for 21 days showed a significant increase (72.8% and 42.3%) in the pancreatic level of TNF-α and IFN-γ, compared to the control saline group (Figures 3 and 4).
On the other hand, treatment of diabetic mice with rosiglitazone (7 mg/kg/day) for 21 days resulted in a significant decrease (29.1% and 39.1%) in the pancreatic level of TNF-α and IFN-γ compared to the diabetic saline group. However, there was a non-significant change compared to the control saline group. Normal mice treated with rosiglitazone (7 mg/kg/day) for 21 days showed a non-significant change in pancreatic TNF-α and IFN-γ content compared to the control saline group (Figures 3 and 4).
 |
|
Figure 3. Effect of rosiglitazone on the TNF-α content in pan-creatic tissue. Mice were treated with rosiglitazone (7 mg/kg/day) for 21 days. Pancreatic TNF-α content was determined at the end of the treatment period. Data are presented as mean ± SEM, n = 13 for diabetic groups and n = 10 for control groups. (*) indicates significant difference from control saline group. (**) indicates significant difference from dia-betic saline group. p < 0.05; ANOVA, Tukey-Kramer test. |
|
 |
|
Figure 4. Effect of rosiglitazone on the IFN-γ content in pancreatic tissue. Mice were treated with rosiglitazone (7 mg/kg/day) for 21 days. Pancreatic IFN-γ content was determined at the end of the treatment period. Data are presented as mean ± SEM, n = 13 for diabetic groups and n = 10 for control groups. (*) indicates significant difference from control saline group. (**) indicates significant difference from diabetic saline group. p < 0.05; ANOVA, Tukey-Kramer test. |
|
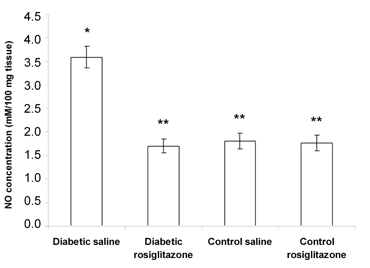
Effect of rosiglitazone on the NO content in pancreatic tissue
Diabetic mice that received saline for 21 days showed a significant increase (98.3%) in the pancreatic NO content compared to the control saline group (Figure 5).
On the other hand, treatment of diabetic mice with rosiglitazone (7 mg/kg/day) for 21 days resulted in a significant decrease (52.6%) in the pancreatic NO content compared to the diabetic saline group. However, there was a non-significant change compared to the control saline group. Normal mice treated with rosiglitazone (7 mg/kg/day) for 21 days showed a non-significant change in the pancreatic NO content compared to the control saline group (Figure 5).
 |
|
Figure 5. Effect of rosiglitazone on the NO content in pancreatic tissue. Mice were treated with rosiglitazone (7 mg/kg/day) for 21 days. Pancreatic NO content (measured as nitrite/nitrate) was determined at the end of the treatment period. Data are presented as mean ± SEM, n = 13 for diabetic groups and n = 10 for control groups. (*) indicates significant difference from control saline group. (**) indicates significant difference from diabetic saline group. p < 0.05; ANOVA, Tukey-Kramer test. |
|
Effect of rosiglitazone on protein expression in pancreatic tissue homogenate
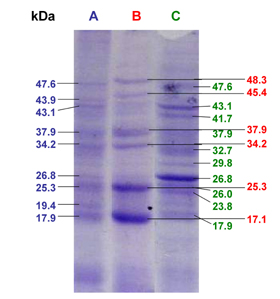
The SDS-PAGE analysis of total protein extracted from pancreatic homogenates is shown in Figure 6 and Table 1.
 |
|
Figure 6. SDS-PAGE analysis of the effect of rosiglitazone on protein expression in pancreatic tissue homogenate from diabetic mice. Mice were treated with rosiglitazone (7 mg/kg) for 21 days. Protein separation of pancreatic tissue homogenate was carried out at the end of the treatment period. A: SDS-PAGE image of pancreatic tissue homogenate of control saline group. B: SDS-PAGE image of pancreatic tissue homogenate of diabetic saline group. C: SDS-PAGE image of pancreatic tissue homogenate of diabetic rosiglitazone group. |
|
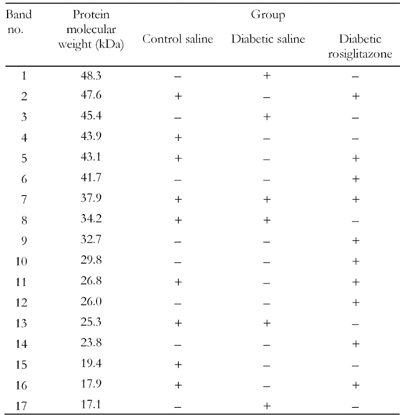
Table
1.
SDS-PAGE analysis of the effect of rosiglitazone on protein expression in pancreatic tissue homogenate from diabetic mice |
|
 |
|
Legend:
+ indicates expression of protein band, - indicates absence of protein band. |
|
There was one specific band at the molecular weight of 37.9 kDa (band no. 7) that was expressed in the pancreatic tissues of normal, diabetic and rosiglitazone-treated diabetic mice.
In diabetic mice, there were three new protein bands at the molecular weights of 48.3, 45.4 and 17.1 kDa which were expressed in the pancreatic tissue compared to normal mice (band nos. 1, 3, 17). However, six bands were expressed at the molecular weights of 47.6, 43.9, 43.1, 26.8, 19.4 and 17.9 kDa in pancreases of normal mice that disappeared in diabetic mice (band nos. 2, 4, 5, 11, 15, 16). Four of these bands were re-expressed after treatment with rosiglitazone (7 mg/kg/day) for 21 days (band nos. 2, 5, 11, 16).
On the other hand, treatment of diabetic mice with rosiglitazone resulted in expression of new protein bands that were not expressed in the pancreases of either normal or diabetic mice. These bands were at the molecular weights of 41.7, 32.7, 29.8, 26, 23.8 kDa (band nos. 6, 9, 10, 12, 14), whereas two bands were expressed at molecular weights of 34.2 and 25.3 kDa (band nos. 8, 13, respectively) that expressed in both normal and diabetic pancreases and again disappeared in mice treated with rosiglitazone.
Histopathological examination to identify the effect of rosiglitazone on pancreatic tissue of MLDSTZ-diabetic mice
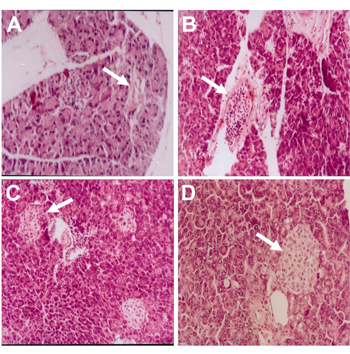
Pancreatic sections from diabetic mice which had received saline for 21 days showed severe hyalinization of pancreatic islets with fibrosis (Figure 7A). Moreover, there was near-complete destruction of the islets with severe (+++) mononuclear cell infiltration (Figure 7B).
In contrast, pancreatic sections from diabetic mice treated with rosiglitazone (7 mg/kg/day) for 21 days showed regenerative pancreatic islets of variable sizes with residual mild (+) mononuclear cell infiltration (Figure 7C). The regenerative islets were well circumscribed and had the same appearance as in the control saline group (Figure 7D).
 |
|
Figure 7. Photomicrograph of H&E-stained pancreatic section from diabetic mice given saline (A, B) for 21 days, showing severe islet hyalinization with fibrosis and near-complete islet destruction with severe (+++) mononuclear cell infiltration, respectively. Mice treated with rosiglitazone (7 mg/kg) for 21 days showed regenerative islets of variable sizes with residual mild (+) mononuclear cell infiltration (C). Pancreatic section of normal mice that received vehicles for 21 days, showing normal islet architecture (D). Magnification: x 250. |
|
Effect of rosiglitazone on immunophenotyping detection of CD4 and CD8 T cells
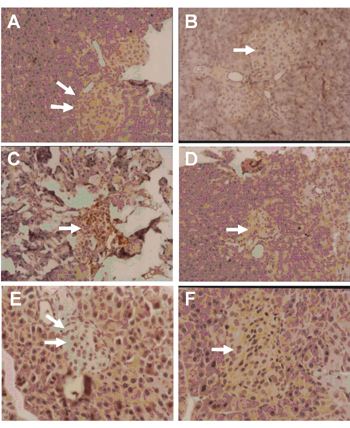
Immunohistochemical staining of pancreatic sections from normal mice which had received saline showed negative staining for both CD4 and CD8 cells infiltrating the pancreatic islets (Figures 8A and 8B).
Pancreatic sections from diabetic mice given saline for 21 days, which were stained with CD4 and CD8 markers, showed dense mononuclear cell infiltrates in which lymphocytes were highly positive for CD4 (Figure 8C) but negative for CD8 stain (Figure 8D).
After treatment of diabetic mice with rosiglitazone (7 mg/kg/day) for 21 days, pancreatic sections stained with CD4 and CD8 markers showed no lymphocytic infiltrate and negative staining for both markers (Figures 8E and 8F).
 |
|
Figure 8. Photomicrograph of CD4 and CD8 immunostaining of pancreatic section from normal mice (A and B), diabetic mice given saline (C and D) or 7 mg/kg rosiglitazone (E and F) for 21 days. A and B show normal pancreatic islets that are negative for CD4- and CD8-stained cells. C shows positive CD4 cells infiltrating islet. D shows fibrotic islet with negative stain for CD8 cells. E and F show regenerated pancreatic islets that are free from both CD4- and CD8-stained cells, respectively. PAP, magnification: x 400. |
|
Discussion
It is well known that there are several hypotheses about the pathogenesis of type 1 DM which involve the immune system playing a major role. In the present study, the experimental model of diabetes induced by CsA/MLDSTZ co-treatment was clearly autoimmune type 1 DM with an immune-mediated component. This was confirmed biochemically and by histopathological examination of the pancreatic tissues of diabetic mice revealing moderate insulitis.
The diabetic model used in this study offered a number of advantages. Firstly, the islet lesions in this model closely resemble those of human disease [28]. Secondly, the mice involved are normal and do not have any underlying immune abnormalities that may complicate the study [29]. Moreover, the onset of diabetes is controlled, so the kinetics of the disease induction may be analyzed [12].
Although rosiglitazone was introduced for the treatment of type 2 DM [19, 20] previous studies demonstrated that rosiglitazone as well as other TZDs could prevent or delay the development of type 1 DM in NOD and MLDSTZ-treated mice [30-32].
In this study, rosiglitazone (7 mg/kg/day) showed marked antidiabetic activity in type 1 DM, as manifested by the significant decrease in the elevated BGL as it reached the normal value at the end of the treatment period.
The reduction in the BGL appeared parallel to a significant increase in the circulating level of insulin after rosiglitazone treatment. The beneficial effect of rosiglitazone on autoimmune type 1 diabetes is confirmed by histopathological findings since pancreatic sections from diabetic mice treated with rosiglitazone showed a significant reduction of mononuclear cell infiltrations in pancreatic islets as well as a regeneration of pancreatic islets which appeared to be well circumscribed.
The beneficial effects of rosiglitazone in this diabetic model may be due to its binding and activation of PPAR-γ expressed on both macrophage [33] and T lymphocytes [34] infiltrating the pancreatic islets during the development of autoimmune diabetes.
PPAR-γ expression is dramatically upregulated in macrophages and T cells during inflammatory responses [35]. As type 1 DM is characterized by infiltration of pancreatic islets with activated macrophages and activated T lymphocytes [2], it may be suggested that rosiglitazone, through activation of these overexpressed PPAR-γ, can inhibit the expression of inflammatory mediators, which participate in the pathogenesis of type 1 DM.
In support of this hypothesis, many natural and synthetic PPAR-γ ligands have been shown to exert anti-inflammatory effects in models of inflammatory bowel disease [36], arthritis [37], ischemia/reperfusion injury [38] and atherosclerosis [39]. The potential anti-inflammatory effects and the clinical benefits of PPAR-γ agonists in these pathological conditions were based on their ability to inhibit transcriptional activation of inflammatory response genes through activation of overexpressed PPAR-γ in activated inflammatory cells [36-39].
In our study, pancreatic levels of both TNF-α and IFN-γ were significantly increased in diabetic mice compared to normal mice. These results were in agreement with previous reports where both TNF-α and IFN-γ genes were expressed in the pancreases of diabetic mice induced by MLDSTZ [12] and in the pancreatic islets isolated from diabetic NOD mice [40]. The synergistic effect of IFN-γ and TNF-α on islet cytotoxicity has been well documented [41-46]. Treatment of diabetic mice with rosiglitazone showed a significant decrease in the pancreatic level of both TNF-α and IFN-γ compared to untreated diabetic mice. These findings were supported by previous reports which documented the fact that, through the activation of PPAR-γ, rosiglitazone could modulate inflammatory processes associated with several inflammatory diseases by reducing the expression of IFN-γ and/or TNF-α [37, 47].
Autoreactive T lymphocytes are considered to be the most important effector cells in autoimmune diabetes. CD4 and CD8 T cells have been shown to be involved in experimental models of type 1 DM, including NOD and MLDSTZ-induced DM [48]. Depletion of either CD4 or CD8 T cell subsets with monoclonal antibodies (mAbs) can prevent NOD or MLDSTZ diabetes, and both subsets are shown to provoke diabetes in NOD mice [48].
In the present study, pancreatic sections from diabetic mice left without treatment for 21 days showed an extensive level of mononuclear cells with positive staining only for CD4 T cells that infiltrated some of the hyalinized pancreatic islets. Similar results showed that CD4 T cells were the only infiltrating cells still present in overtly diabetic animals [40]. CD8 T cells were not observed in the islets at this stage [40]. Treatment of diabetic mice with rosiglitazone caused regeneration of pancreatic islets without any CD4 and CD8 T cell infiltration. The disappearance of CD4 T cells after rosiglitazone treatment could be explained by the inhibitory effect of rosiglitazone on T lymphocytes through the activation of PPAR-γ. This inhibitory effect resulted in the decrease in TNF-α and IFN-γ levels after treatment which, in turn, inhibited the activation and migration of further inflammatory cells including CD4 T cells into the islets. Through activation of PPAR-γ, rosiglitazone may inhibit CD8 T-cells infiltrating the islet during the early stages of diabetes [40] and this effect could also be considered as a possible mechanism underlying the beneficial effect of rosiglitazone on type 1 DM.
In animal models of type 1 DM, the role of NO in β-cell destruction has been clearly demonstrated [49]. In the present study, the pancreatic level of NO in diabetic mice was significantly increased compared to normal mice. This was in agreement with several reports suggesting that NO production contributed to the cytotoxicity of MLDSTZ to pancreatic β-cells [14, 15, 50]. In MLDSTZ-induced diabetes, it was found that the incidence of hyperglycemia was decreased when iNOS inhibitors were used [14]. In addition, it was found that transgenic mice deficient in iNOS have reduced sensitivity to MLDSTZ-induced diabetes [15]. In our study, the decrease in the NO level in pancreatic homogenate from diabetic mice given rosiglitazone may result from the following mechanisms. Firstly, direct activation of PPAR-γ expressed on the pancreatic β-cell [51] induced a decrease in iNOS expression and NO production inside the β-cell. Secondly, the decrease in TNF-α and IFN-γ after rosiglitazone treatment may result in inhibition of NO production from both β-cell and macrophages infiltrating the pancreatic islets. Thirdly, rosiglitazone was found to induce apoptosis of macrophages activated with TNF-α and IFN-γ [52]. The death of macrophages could in turn result in a decrease in the level of its inflammatory mediators, including NO and TNF-α.
In type 1 DM it has been suggested that β-cell-selective toxic effects of cytokines and free radicals might be reflected at the level of islet protein expression [53, 54]. In this context, the expressions of rat islet proteins were found to be changed after IL-1 exposure [53, 55, 56]. Protein synthesis inhibitors such as cycloheximide were found to protect IL-1-exposed islets from destruction [56].
In the present study, the different pattern of protein expression observed in the pancreas of diabetic mice could be a secondary consequence of the elevated pancreatic levels of TNF-α, IFN-γ and NO. The differentially expressed bands between the pancreases of normal and diabetic mice may be related to functional or structural proteins associated with β-cell damage and dysfunction. In addition, the protein bands that changed after rosiglitazone treatment may be those of some inflammatory mediators expressed in the pancreas of diabetic mice, and this expression is documented as being decreased by PPAR-γ activators.
Islets cells have been documented as expressing significantly high levels of PPAR-γ [51]. It may therefore be suggested that rosiglitazone might act directly on pancreatic islet cells, resulting in changes in protein expression as well as regeneration of the destroyed pancreatic islets, as could also be observed in our study. In support of this suggestion, it was reported that nine proteins were differentially expressed between the pancreatic tissue of lep/lep diabetic mice and lean littermates [57]. Four of these proteins were altered toward their level in the lean littermates after rosiglitazone treatment [57]. Similarly, previous studies showed that rosiglitazone modulated the expression of different proteins in various tissues, which may be an underlying factor in its therapeutic effect [58].
In conclusion, our study showed that rosiglitazone may be effective in the treatment of autoimmune DM. The antidiabetic effect of rosiglitazone seemed to be a secondary consequence of its anti-inflammatory and immunomodulatory properties, as it resulted in a significant decrease in the elevated levels of TNF-α, IFN-γ and NO. The pancreatic islets of mice receiving rosiglitazone were nearly free from CD4 and CD8 lymphocytes. In addition, the antidiabetic effects of rosiglitazone might be reflected at the molecular level of islet protein expression.
References
- Gepts W, In't Veld PA. Islet morphological changes. Diabetes Metab Rev 1987. 3:859-872. [DOD]
- Rabinovitch A. Role of cell-mediated immunity and cytokines in the pathogenesis of type 1 diabetes mellitus. In: Le Roith D, Taylor SI, Olefsky JM. Diabetes Mellitus a Fundamental and Clinical Text 2000. pp 383-398. [DOD]
- Rabinovitch A, Suarez-Pinzon WL. Role of cytokines in the pathogenesis of autoimmune diabetes mellitus. Rev Endocr Metab Disord 2003. 4:291-299. [DOD] [CrossRef]
- Major CD, Gao ZY, Wolf BA. Activation of the sphingomyelinase/ceramide signal transduction pathway in insulin-secreting beta-cells: role in cytokine induced beta-cell death. Diabetes 1999. 48:1372-1380. [DOD]
- Lehmann PV, Sercerz EE, Forsthuber T, Dayan CM, Gammon G. Determinant spreading and the dynamics of the autoimmune T cell repertoire. Immunol Today 1993. 14:203-207. [DOD] [CrossRef]
- Faust A, Rothe H, Schade U, Lampeter E, Kolb H. Primary nonfunction of islet grafts in autoimmune diabetic nonobese diabetic mice is prevented by treatment with interleukin-4 and interleukin-10. Transplantation 1996. 62:648-652. [DOD] [CrossRef]
- Suarez-Pinzon W, Sorensen O, Bleackley RC, Elliott JF, Rajotte RV, Rabinovitch A. Beta-cell destruction in NOD mice correlates with Fas (CD95) expression on beta-cells and proinflammatory cytokines expression in islets. Diabetes 1999. 48:21-28. [DOD]
- Like AA, Rossini AA. Streptozotocin induced pancreatic insulitis: new model of diabetes mellitus. Science 1976. 193:27-54. [DOD] [CrossRef]
- Kolb H, Kroncke KD. Lesson from the low-dose streptozotocin model in mice. Diabetes Rev 1993. 1: 16-26. [DOD]
- Roep BO, Atkinson M, von Herrath M. Satisfaction (not) Guaranteed: re-evaluating the use of animal models of type 1 diabetes. Nat Rev Immunol 2004. 4:989-997. [DOD] [CrossRef]
- Snadberg JO, Anersson A, Eizirik DL, Sandler S. Interleukin-1 receptor antagonist prevents low dose streptozotocin induced diabetes in mice. Biochem Biophys Res Commun 1994. 202:543-548. [DOD] [CrossRef]
- Herold KC, Vezys V, Sun Q, Viktora D, Seung E, Reiner S, Brown D. Regulation of cytokine production during development of autoimmune diabetes induced with multiple low doses of streptozotocin. J Immunol 1996. 156:3521-3527. [DOD]
- Holdstad M, Sandler S. A transcriptional inhibitor of TNF-alpha prevents diabetes induced by multiple low-doses streptozotocin injections in mice. J Autoimmun 1996. 156:3521-3527. [DOD]
- Kolb H, Kicscel U, Kroncke KD, Kolb-Bachofen V. Suppression of low-dose streptozotocin induced diabetes in mice by administration of nitric oxide synthase inhibitor. Life Sci 1991. 49:L213-L217. [DOD] [CrossRef]
- Flodstrom M, Tyrberg B, Eizirik DL, Sandler S. Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes 1999. 48:706-713. [DOD]
- Cvetkovic I, Al-Abed Y, Miljkovic D, Maksimovic-Ivanic D, Roth J, Bacher M, Lan H, Nicoletti F, Stosic-Grujicic S. Critical role of macrophage migration inhibitory factor activity in experimental autoimmune diabetes. Endocrinology 2005. 146(7):2942-2951. [DOD] [CrossRef]
- Fraser RB, Rowden G, Colp P, Wright JR. Immunophenotyping of insulitis in control and essential fatty acid deficient mice treated with multiple low-dose streptozotocin. Diabetologia 1997. 40:1263-1268. [DOD] [CrossRef]
- Wright JR, Fraser RB, Kapoor S, Cook HW. Essential fatty acid deficiency prevents multiple low-dose streptozotocin-induced diabetes in naive and cyclosporine-treated low-responder murine strains. Acta Diabetologica 1995. 32:125-130. [DOD] [CrossRef]
- Kahn CR, Chen L, Cohen SE. Unraveling the mechanism of action of thiazolidinedione. J Clin Invest 2000. 106:1305-1307. [DOD]
- Noble J, Baerlocher MO, Silverberg J. Management of type 2 diabetes mellitus. Role of thiazolidinediones. Can Fam Physician 2005. 51:683-687. [DOD]
- Desvergne B and Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocrine Reviews 1999. 20:649-688. [DOD] [CrossRef]
- Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol 2001. 169:453-459. [DOD] [CrossRef]
- Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Maffia P, Patel NS, Di Paola R, Ialenti A, Genovese T, Chatterjee PK, et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-gamma, reduces acute inflammation. Eur J Pharmacol 2004. 483(1):79-93. [DOD] [CrossRef]
- Hasegawa H, Takano H, Zou Y, Qin Y, Hizukuri K, Odaka K, Toyozaki T, Komuro I. Pioglitazone, a peroxisome proliferator-activated receptor gamma activator, ameliorates experimental autoimmune myocarditis by modulating Th1/Th2 balance. J Mol Cell Cardiol 2005. 38(2):257-265. [DOD] [CrossRef]
- Trinder P. Determination of blood glucose using an oxidase-peroxidase system with a non-carcinogenic chromogen. J Clin Pathol 1969. 22(2):158-161. [DOD]
- Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 2001. 5(1):62-71. [DOD] [CrossRef]
- Laemmli AS. Cleavage of structural protein during the assembly of the deep bacterophage CT4. Nature 1970. 227:680-684. [DOD] [CrossRef]
- Somoza N, Vargas F, Roura-Mir C, Vives-Pi M, Fenandez Figueras MT, Ariza A, Gomis R, Bragado R, Marti M, Jaraquemada D, Pujol-Borrell R. Pancreas in recent onset insulin-dependent diabetes mellitus. J Immunol 1994. 153:1360-1377. [DOD]
- Serreze DV, Gaskins HR, Leiter EH. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J Immunol 1993. 150:2534-2543. [DOD]
- Beales PE, Liddi R, Giorgini AE, Signore A, Procaccini E, Batchelor K, Pozzilli P. Troglitazone prevents insulin dependent diabetes in the non-obese diabetic mouse. Eur J Pharmacol 1998. 357:221-225. [DOD] [CrossRef]
- Beales PE and Pozzilli P. Thiazolidinediones for the prevention of diabetes in non-obese dependent (NOD) mouse: implications for human type 1 diabetes. Diabetes Metab Res Rev 2002. 18:114-117. [DOD] [CrossRef]
- Takamura T, Andro H, Nagai Y, Yamashita H, Nohora E, kobayashi K. Pioglitazone prevents mice from multiple low-dose streptozotocin-induced insulitis and diabetes. Diabetes Res Clin Pract 1999. 44 (2):107-114. [DOD] [CrossRef]
- Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. PNAS 2003. 100(11):6712-6717. [DOD] [CrossRef]
- Wang YL, Frauwirth KA, Rangwala SM, Lazar MA, Thompson CB. Thiazolidinedione activation of Peroxisome proliferator-activated receptor γ can enhance mitochondrial potential and promote cell survival. J Biol Chem 2002. 277(35):31781-31788. [DOD] [CrossRef]
- Daynes RA and Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2002. 2:748-759. [DOD] [CrossRef]
- Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer: a basis for new therapeutic strategies. J Exp Med 2001. 193:827-838. [DOD] [CrossRef]
- Cuzzocrea S, Mazzon E, Dugo L, Patel NS, Serraino I, Di Paola R, Genovese T, Britti D, De Maio M, Caputi AP, et al. Reduction in the evolution of murine type II collagen-induced arthritis by treatment with rosiglitazone, a ligand of the peroxisome proliferator-activated receptor gamma. Arthritis Rheum 2003. 48(12):3544-3556. [DOD] [CrossRef]
- Ito H, Nakano A, Kinoshita M, Matsumori A. Pioglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates myocardial ischemia/reperfusion injury in a rat model. Lab Invest 2003. 83(12):1715-1721. [DOD] [CrossRef]
- Marx N, Kehrle B, Kohlhammer K Grub M, Koenig W, Hombach V, Libby P, Plutzky J. PPAR activators as anti-inflammatory mediators in human T lymphocytes: implication for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res 2002. 90:703-710. [DOD] [CrossRef]
- Papaccio G, Ammendole E, Pisanti FA. Nicotinamide decreases MHC class II but not MHC class I expression and increases intercellular adhesion molecule-1 structures in non-obese diabetic mouse pancreas. J Endocrinol 1999. 160:389-400. [DOD] [CrossRef]
- Campbell IL, Oxbrow L, Harrison LC. Interferon-gamma: pleiotropic effects on a rat pancreatic beta cell line. Mol Cell Endocrinol 1987. 52:161-167. [DOD] [CrossRef]
- Defilippi P, Poupart P, Tavernier J, Fiers W, Contant W. Induction and regulation of mRNA encoding 26-kDa protein in human cell line treated with recombinant human tumor necrosis factor. Proc Natl Acad Sci USA 1987. 84:4557-4561. [DOD] [CrossRef]
- Dealtry GB, Naylor MS, Fiers W, Balkwill FR. DNA fragmentation and cytotoxicity caused by tumour necrosis factor is enhanced by interferon-gamma. Eur J Immunol 1987. 17:689-693. [DOD]
- Campbell IL, Iscaro A, Harrison LC. IFN-gamma and tumor necrosis factor-alpha: cytotoxicity to murine islets of langerhans. J Immunol 1988. 141:2325-2329. [DOD]
- Campbell IL, Wong GH, Schrader JW, Harrison LC. Interferon-gamma enhances the expression of the major histocompatibility class 1 antigens on mouse pancreatic beta cells. Diabetes 1985. 34:1205-1209. [DOD]
- Campbell IL, Oxbrow L, West J, Harrison LC. Regulation of MHC protein expression in pancreatic beta cells by interferon-gamma and tumour necrosis factor-alpha. Mol Endocrinol 1988. 2:101-107. [DOD]
- Hong G, Davis B, Khatoon N, Baker SF, Brown J. PPAR gamma-dependent anti-inflammatory action of rosiglitazone in human monocytes: suppression of TNF alpha secretion is not mediated by PTEN regulation. Biochem Biophys Res Commun 2003. 303(3):782-787. [DOD] [CrossRef]
- Miller BJ, Appel MC, O'Neil J, Wicker LS. Both the LYT-2+ and L3T4+ T cell subsets are required for transfer of diabetes in non obese diabetic mice. J Immunol 1988. 140:52-58. [DOD]
- Thomas HE, Darwiche R, Corbett JA, Kay TW. Interleukin-1 plus gamma-interferon-induced pancreatic beta-cell dysfunction is mediated by beta-cell nitric oxide production. Diabetes 2002. 51(2):311-316. [DOD]
- Fehsel K, Kroncke KD, Kolb-Bachofen V. The action of NO and its role in autoimmune diabetes mellitus. Res Immunol 1995. 146:711-715. [DOD] [CrossRef]
- Dubois M, Pattou F, Kerr-Conte J, Vandewalle B, Desreumaux P, Auwerx J, Schoonjans K, and Lefebvre J. Expression of peroxisome proliferators-activated receptor gamma (PPARgamma) in normal human pancreatic islet cells. Diabetologia 2000. 43:1165-1169. [DOD] [CrossRef]
- Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, Fruchart JC, Chapman J, Najib J. Activation of proliferators-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem 1998. 273(40):25573-22580. [DOD] [CrossRef]
- Andersen HU, Larsen PM, Fey SJ, Karlsen AE, Mandrup-Poulsen T, Nerup J. Two-dimensional gel electrophoresis of rat islets proteins: interleukin-1 beta induced changes in protein expression are reduced by L-arginine depletion and nicotinamide. Diabetes 1995. 44:400-407. [DOD]
- Karlsen AE, Andersen HU, Vissing H, Mose Larsen P, Fey SJ, Cuartero BG, Madsen OD, Petersen JS, Mortensen SB, Mandrup-Poulsen T, et al. Cloning and expression of cytokine inducible nitric oxide synthase cDNA from rat islets of Langerhans. Diabetes 1995. 44:753-758. [DOD]
- Larsen PM, Fey SJ, Larsen MR, Nawrocki A, Andersen HU, Kahler H, Heilmann C, Voss MC, Roepstorff P, Pociot F, et al. Proteome analysis of interleukin-1beta-induced changes in protein expression in rat islets of langerhans. Diabetes 2001. 50:1056-1063. [DOD]
- Eizirik DL, Bjorklund A, Welsh N. Interleukin-1-induced expression of nitric oxide synthase in insulin producing cells is preceded by c-fos induction and depends on gene transcription and protein synthesis. FEBS Lett 1993. 317:62-66. [DOD] [CrossRef]
- Sanchez JC, Converset V, Nolan A, Schmid G, Wang S, Heller M, Sennitt MV, Hochstrasser F, Cawthorne MA. Effect of rosiglitazone on the differential expression of diabetes-associated proteins in pancreatic islets of C57Bl/6 lep/lep mice. Molecular and Cellular Proteomics 2002. 1:509-516. [DOD] [CrossRef]
- Rieusset J, Chambrier C, Bouzakri K, Dusserre E, Auwerx J, Riou JP, Laville M, Vidal H. The expression of the p85alpha subunit of phosphatidylinositol 3-kinase is induced by activation of the peroxisome proliferator-activated receptor gamma in human adipocytes. Diabetologia 2001. 44:544-554. [DOD] [CrossRef]
This article has been cited by other articles:

|
Differential Transcriptional Expression of PPARalpha, PPARgamma1, and PPARgamma2 in the Peritoneal Macrophages and T-cell Subsets of Non-obese Diabetic Mice
Yaacob NS, Kaderi MA, Norazmi MN
J Clin Immunol 2009. 29(5):595-602
|
|

|
Rosiglitazone Modulates Insulin-Induced Plasma Membrane Area Changes in Single 3T3-L1 Adipocytes
Velebit J, Kovacic PB, Prebil M, Chowdhury HH, Grilc S, Kreft M, Jensen J, Isenovic ER, Zorec R
J Membr Biol 2008. 223(3):141-149
|
|
|
Interaction among abnormal fatty acid oxidation, endothelial function disorder, and oxidative stress in the onset of severe preeclampsia
Wang JL, Yang Z, Wang R, Zhu JM
Zhonghua Yi Xue Za Zhi (National Medical Journal of China) 2008. 88(21):1471-1475
|
|

|
Transcription factors FoxO3 a expression on adipose tissue of KKay diabetic mice and the effects of treatment with rosiglitazone and metformin
Li YX, Zeng JB, Sun Q, Wang H
Basic and Clinical Medicine 2008. 28(3):260-263
|
|

|
Modulatory Effects of 6-Carboxymethylthiopurine on Activated Murine Macrophages
De Paula ML, Braga FG, Coimbra ES, Carmo AM, Teixeira HC, Da Silva AD, Souza MA, Ferreira AP
Chem Biol Drug Des 2008. 71(6):563-567
|
|
|


)
)
)
)
)
)
)
)
)

