Editorial
| Rev Diabet Stud,
2007,
4(4):200-209 |
DOI 10.1900/RDS.2007.4.200 |
TNFalpha in the Pathogenesis of Diabetes-Induced Embryopathies: Functions and Targets
Arkady Torchinsky, Vladimir Toder
Department of Cell and Developmental Biology, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
Address correspondence to: Arkady Torchinsky, e-mail: arkadyt@post.tau.ac.il
Manuscript submitted January 21, 2008; resubmitted February 22, 2008; accepted February 25, 2008.
Keywords: diabetes, embryopathy, pregnancy loss, inborn anomalies, TNFalpha, NF-kappaB, LIF
Abstract
Hyperglycemia-induced increase in the production of reactive oxygen species (ROS) is proposed to be an initial step in the pathogenesis of diabetes-induced spontaneous abortions and structural inborn anomalies. However, the subsequent steps in this process are incompletely understood. One of the key molecules involved is tumor necrosis factor-alpha (TNFα): its expression is regulated by ROS and it regulates ROS production in turn. This cytokine has been the focus of many studies addressing the mechanisms of different forms of diabetes-induced embryopathies, such as early pregnancy loss, inborn anomalies, fetal growth retardation as well as some pathologies appearing during adult life. In this review, we analyze the results of these studies and discuss how TNFα may regulate the response of pre- and post-implantation stage embryos to diabetes-induced detrimental stimuli. The data presented in this review suggest that TNFα may play a dual role in the pathogenesis of diabetes-induced embryopathies. It may act both as a mediator of diabetes-induced embryotoxic stimuli leading to the death of peri-implantation stage embryos and, possibly, as a suppressor of diabetes-induced apoptosis in post-implantation stage embryos. It also appears that TNFα fulfills these functions via interaction with leukemia inhibitory factor (LIF) and the transcription factor NF-κB. These molecules are presently considered as attractive targets for the treatment of diabetes-induced complications. Therefore, further studies addressing their role in the mechanisms underlying diabetes-induced embryopathies are needed to evaluate the safety of such therapies for diabetic women of childbearing age.
Introduction
Spontaneous abortions and structural inborn anomalies are the main complications of diabetic pregnancy [1]. Early epidemiological observations revealed that the incidence of these adverse pregnancy outcomes correlates with high blood glucose and glycosylated hemoglobin (HbA1c) levels [2-4]. Rigorous glycemic control can reduce the rate of these complications in diabetic pregnancies [5], but the rate is still three to fivefold greater than for non-diabetic pregnancies [6].
In recent years, there has been considerable progress in our understanding of the etiology and pathogenesis of diabetes-induced embryopathies. Until recently, the role of glucose as a causative factor for diabetes-induced inborn anomalies remained questionable. About 30 years ago, Ornoy and Cohen were the first to suggest that glucose may have a teratogenic effect in vivo [7]. Later, the hypothesis was developed that glucose is not teratogenic per se but rather a marker for other diabetes-generated teratogens such as ketone bodies, triglycerides, branched chain amino acids, sorbitol or glycated proteins [8, 9]. In our in vivo study, we demonstrated that the occurrence of severely malformed fetuses in litters of streptozotocin (STZ)-induced diabetic ICR mice is associated with glucose levels >27.8 mmol/l [10]. This result appeared to support the latter hypothesis. On the other hand, this study did not detract from the suggestion made by Ornoy and Cohen that episodes of elevated glucose levels can induce congenital anomalies. The reason is that malformed fetuses were observed also in litters of STZ-treated mice, which were considered to be non-diabetic at the end of pregnancy [7]. Recently, a study performed by Loeken's team provided convincing evidence that elevated glucose levels are the main etiological factor for diabetes-induced inborn anomalies [11].
Most researchers now accept that hyperglycemia-induced increase in the production of reactive oxygen species (ROS) is an initial key event in the pathogenesis of diabetes-induced structural anomalies [12-15]. However, as ROS are capable of regulating numerous intracellular signal transduction pathways [16], subsequent pathological events seem to be far from completely understood. One of the key molecules involved is tumor necrosis factor-alpha (TNFα): its expression is regulated by ROS and it regulates ROS production in turn [17]. This cytokine has been the focus of many studies addressing the mechanisms underlying diabetes-induced embryopathies [18, 19]. In this review, we analyze the results of these studies and discuss how and via which targets TNFα may regulate the response of pre- and post-implantation stage embryos to diabetes-induced detrimental stimuli.
TNFα and diabetes-induced early pregnancy loss
TNFα mediates diabetes-induced death of early embryos
Experiments in STZ- or alloxan (ALX)-induced diabetic female mice [10, 20, 21] or rats [22] showed that their pregnancy rate (the proportion of mated females that become pregnant) is significantly lower than that of their non-diabetic counterparts. In these studies, neither implantation sites nor resorptions were found in the uteri of mated diabetic but non-pregnant females. These results suggested that pregnancy failure in such females resulted from diabetes-induced early embryonic death, i.e. death of the pre- or peri-implantation stage embryos. To clarify this question, we evaluated the pregnancy rate in STZ-induced diabetic mice on days 4 (the end of the pre-implantation period) and 8 (the end of the implantation period) of pregnancy [23]. We found that the pregnancy rate was identical in diabetic and non-diabetic females tested on day 4, but not on day 8, of pregnancy. On day 8 of pregnancy, diabetic females exhibited a decrease in the pregnancy rate. We concluded that pregnancy failure in those mice resulted from death of peri-implantation stage embryos.
Our study also revealed that the pregnancy rate in STZ-induced diabetic TNFα knockout mice was lower than that in non-diabetic females but far higher than that recorded in their TNFα-positive counterparts [23]. The critical outcome of this observation was that the cytokine may be a central component in the mechanisms underlying diabetes-induced death of early embryos.
Does TNFα mediate the diabetes-induced death of peri-implantation embryos by affecting the embryo or uterus?
Based on the fact that diabetes-induced early pregnancy loss results from death of peri-implantation embryos, two scenarios mediated by TNFα are conceivable. Either the cytokine affects the ability of pre-implantation stage embryos to implant in the uterus or it impairs the ability of the uterus to establish implantation.
The first scenario is supported by data demonstrating the ability of TNFα to affect early embryos adversely. Indeed, it has been observed that rat blastocysts exposed to TNFα exhibited decreased cell proliferation [24] and an increased rate of blastomere apoptosis [25]. The same impairments have been observed in cultured mice and cattle blastocysts exposed to TNFα [26, 27]. The relevance of these findings to maternal diabetes was shown in a study by Pamper and coworkers [28]. In this study, the authors incubated rat blastocysts in a diabetic culture medium and found an improved growth when pretreating the blastocysts with anti-sense oligonucleotides that blocked TNFα receptors. It is also worth noting that suppression of cell proliferation and excessive apoptosis were observed in pre-implantation mouse and rat embryos developing in a diabetic environment (references in [29]).
Although the above mentioned data show that TNFα can injure early embryos, a scenario, in which these injures are held responsible for early pregnancy loss in diabetic females, seems questionable. There is a considerable inter- and intra-litter variability in the susceptibility of embryos to detrimental stimuli [30]. If TNFα-induced injures in embryos were responsible for early pregnancy loss, then we would observe a significant decrease in the implantation rate in pregnant diabetic females at any time point after completing the implantation period. Although three studies found lower implantation rates [22, 31, 32], in most studies the rate did not differ from that observed in non-diabetic females [7, 10, 20, 21, 23, 33-43]. When blastocysts were exposed to diabetic environments or TNFα, the cellular deficit in these blastocysts was mostly at the expense of the inner cell mass (ICM) - the cells that form the fetus - but not of the trophectoderm (TE) cells that ensure implantation of the blastocyst into the uterine wall [44]. Consistent with this finding, embryo transfer experiments revealed that TNFα-treated mouse blastocysts implant practically at the same rate as control blastocysts but exhibit a higher incidence of resorptions (i.e. post-implantation death) [29]. The same result was obtained in an embryo transfer study in non-obese diabetic (NOD) mice. While the implantation rate of NOD blastocysts transferred to the uteri of ICR mice (control) did not differ from that of ICR embryos transferred to ICR uteri, the level of resorptions of the former was significantly higher than that of the latter [45]. In the light of these findings, it seems reasonable to suggest that diabetes-induced alterations in pre-implantation embryos mainly disturb their ability to develop rather than implant into the uterus.
The second possible scenario explaining early pregnancy loss proposes that the death of peri-implantation embryos in diabetic females results from TNFα-induced injuries in the peri-implantation uterus. This view is based on data demonstrating that TNFα mRNA and protein are overexpressed in the uterine cells of diabetic females from the initiation of implantation and onwards ([42] and references in [18, 19]). As TNFα is able to activate the death receptor-mediated apoptotic signaling cascade [46], these observations suggest that the apoptotic process in the uterus, which is crucial for the implantation of the embryo [47], may be distorted in diabetic mice. The suggestion is supported by studies showing that mouse uterine epithelial WEG-1 and human endometrial HEC-1 cells exposed to TNFα or cultured in a diabetic condition exhibit a decreased viability and several apoptotic markers [18]. A mechanistic role for the uterus in diabetes-induced early pregnancy loss is also suggested by data demonstrating a lower implantation rate of ICR blastocysts (control) transferred to NOD uteri compared with that of ICR blastocysts transferred to ICR uteri [45]. Finally, this scenario may explain why the death of peri-implantation embryos in some diabetic females is not accompanied by a decrease in the implantation rate in diabetic females who retain pregnancy.
In summary, the observations mentioned above appear to suggest that the peri-implantation uterus rather than the pre- or peri-implantation embryo itself is the target and that TNFα may play a role in mediating diabetes-induced early pregnancy loss. This view is further discussed in the following section.
Is the TNFα-induced apoptosis in the uterus a mechanism of diabetes-induced early pregnancy loss?
For successful implantation, the temporal pattern and intensity of apoptosis in the peri-implantation uterus have to be tightly regulated [47]. Therefore, it is conceivable that TNFα-induced death receptor-mediated activation of apoptosis in the peri-implantation uterus may be harmful to implantation. However, it is important to bear in mind that TNFα-activated death receptor-mediated signaling cascade also activates the transcription factor NF-κB [48], which is a powerful negative regulator of apoptosis. Considerable evidence suggests that its activation protects cells against TNFα-induced apoptosis [49, 50]. A recent study in a mouse model of autoimmune type 1 diabetes demonstrated the anti-apoptotic role of NF-κB in pancreatic β-cells exposed to TNFα [51].
As to the role of NF-κB localized in the uterus, lack of activity on its part does not seem to impair implantation, as was observed in experimental mice where NF-κB activation has been blocked (references in [52]). At the same time, NF-κB activity in uterine cells is tightly regulated during implantation [53]. Furthermore, NF-κB was found to be activated in uterine cells exposed to TNFα and it was suggested that this event might be a transient NF-κB-dependent anti-apoptotic reaction [18].
In summary, excessive apoptosis in the peri-implantation uterus cannot be excluded as a possible cause for diabetes-induced early embryonic death. Nor can we exclude the possibility that TNFα-activated death receptor-mediated signaling in uteri of diabetic females retaining pregnancy may be an event supporting implantation via the activation of NF-κB-mediated anti-apoptotic signaling. In the latter case, it is possible that TNFα mediates diabetes-induced early pregnancy loss by affecting mechanisms that regulate uterine receptivity for blastocyst implantation. We discuss these mechanisms in the next section.
LIF as a possible TNFα target
One of the main mechanisms ensuring uterine receptivity is associated with the function of leukemia inhibitory factor (LIF) [54]. Earlier, we hypothesized that LIF may be involved in pathways underlying stress-induced TNFα-mediated early pregnancy loss [55]. In this section, we discuss this hypothesis in more detail.
The level of LIF in the uterus is tightly regulated, reaching a peak at the time of implantation [54]. In mice, implantation does not occur in LIF-/- uteri, whereas LIF null blastocysts develop successfully to term in wild-type females [56]. LIF is able to trigger several signaling pathways, including the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway, which is currently suggested to be essential for the acquisition of uterine receptivity [54, 57]. LIF triggers this pathway by binding to its receptors (LIFR), followed by activation of the JAK family kinases, which, in turn, activate STATs transcription factors, in particular STAT-3 [57]. The importance of STAT-3 for the acquisition of uterine receptivity has been demonstrated [58] and it is now known that the JAK/STAT signaling pathway must be activated for successful implantation [54].
TNFα is able to induce LIF expression in many cell types, including human endometrial epithelial and stromal cells in a concentration and time-dependent manner (references in [55]). TNFα is also a powerful activator of NF-κB, which, in turn, activates TNFα expression [49, 50]. It has been demonstrated that the promoter region of the LIF gene contains an NF-κB binding site [59] and that NF-κB may mediate TNFα-stimulated LIF production in human endometrial epithelial cells [60]. NF-κB was also suggested to be involved in the regulation of STAT-3 DNA-binding [57]. Based on these data, we may propose a model, in which TNFα overexpression alters the function of LIF in the uterus of diabetic mice leading to the death of peri-implantation embryos (Figure 1).
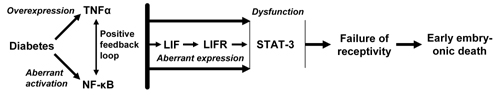 |
 |
Figure 1. A model of a pathway triggered by TNFα in the uterus of diabetic mice, which can lead to the death of peri-implantation embryos. Diabetes increases TNFα expression and activates NF-κB in the uterus. These effects may be boosted via a positive feedback loop between these molecules and followed by TNFα- and/or NF-κB-induced dysregulation of molecules, such as LIF, LIFR and STAT-3, which have a critical function for uterine receptivity. |
|
Yet, there are still many unanswered questions. One question is whether diabetes-induced TNFα-mediated pregnancy loss may result from an increase or decrease in LIF production in the uterus. Indeed, some increase in TNFα and decrease in LIF expression were observed in fluid derived from uterine irrigation of women with recurrent failed embryo transfer [61]. At the same time, data exist suggesting that LIF overexpression in the uterine lumen may be also harmful to implantation [62]. The condition is complicated by the complex organization of the LIF gene that is involved in translation of intracellular and extracellular proteins with distinct cellular activities [63]. Furthermore, it is possible that the direction of the LIF secretion by polarized uterine epithelium may be a factor determining the outcome of implantation. It has been suggested that secretion towards the basal cells is necessary for implantation, whereas secretion in the apical direction towards the uterine lumen may be harmful for implantation [62].
In addition, it is unclear, whether LIF-mediated regulation of the implantation rate of mouse and rat embryos is an "all-or-nothing" phenomenon. The answer to this question is important because it will make it possible to estimate the extent to which other factors regulating uterine receptivity may be involved in mechanisms underlying diabetes-induced early pregnancy loss.
Finally, TNFα does not seem to be the only molecule capable of affecting the function of LIF in the diabetic uterus. In an experimental study, the gene encoding LIF was found to be a potential target for the tumor suppressor gene p53 [64]. The authors observed that the dramatic decrease in pregnancy rate in p53-/- female mice, as compared to that recorded in p53+/+ females, was accompanied by a decrease in uterine LIF mRNA expression. Although there is no proof for changes in p53 expression in uterine cells of diabetic mice, studies demonstrating that p53 is activated in the process of hyperglycemia-induced apoptosis in pre-implantation stage embryos [65] suggest that such changes are possible. In the light of evidence that not only decreased but also increased production of LIF in the uterus may be harmful for implantation [62], it is important to investigate the role of p53 as a possible regulator of LIF secretion in the peri-implantation uterus of diabetic females.
TNFα and diabetes-induced inborn structural anomalies
Targets for diabetes-induced teratogenic injuries
The possibility that the hyperglycemia-induced injury of pre-implantation embryos may predispose them to malformations later in embryogenesis [66] or even directly result in structural malformations [67] can not be excluded. Studies using whole embryo culture suggest that diabetes-induced neural tube defects (NTDs) (exencephaly, anencephaly, microcephaly, spina bifida) result from a hyperglycemia-induced injury of gastrulation and neurulation-stage embryos [68, 69]. It has been proposed that alterations in the histiotrophic function of the visceral yolk sac (VYS) may also be involved in the pathogenesis of hyperglycemia-induced malformations in these embryos [70]. However, subsequent studies [69] have failed to confirm a relationship between altered VYS hystotrophic function and the occurrence of structural anomalies. Presently available data suggest that the embryo at the neural tube closure stage (between 8.5 and 10.5 embryonic days in mice) is the main target of the diabetes-induced teratogenic stimuli.
A possible role for TNFα in diabetes teratogenesis
Evidence for the role of TNFα diabetes-induced malformations in embryos was generated in experimental studies with diabetic TNFα knockout mice. In these studies, the proportion of malformed fetuses in diabetic TNFα+/+ mice was lower than in diabetic TNFα-/- mice [23]. On the assumption that TNFα acts as a mediator of diabetes-induced death of peri-implantation stage embryos, the most acceptable explanation for this phenomenon is that death decreases the proportion of teratologically sensitive TNFα+/+ embryos in a population exposed to a diabetes-induced teratogenic effect taking place after implantation. However, the number of implantation sites in diabetic TNFα+/+ mice was practically identical to that in non-diabetic TNFα+/+ mice.
Another explanation for TNFα-induced malformations might be associated with an increased death of post-implantation TNFα+/+ embryos with severe structural anomalies. However, we observed that the number of living fetuses in diabetic TNFα+/+ mice did not differ significantly from that in TNFα-/- mice. Together, these results implied that TNFα-mediated mechanisms aimed at preventing the formation of diabetes-induced structural anomalies may operate in post-implantation embryos.
Possible mechanisms underlying the TNFα-regulated response to diabetes-induced teratogenic injury
It has been proposed that diabetes-induced NTDs arise from incomplete closure of the neural tube. For the neural tube to be formed, the apoptotic process involved in its formation has to be tightly regulated [71]. A large number of studies demonstrate excessive apoptosis in the brain of early post-implantation stage embryos that develop in a diabetic environment and exhibit open neural folds [38, 72-75]. Interestingly, excessive apoptosis was also observed in the VYS of these embryos [76]. While the role of excessive apoptosis in the VYS in the pathogenesis of diabetes-induced NTDs remains unclear, excessive apoptosis in the embryonic brain appears to be mechanistically linked to this pathology. Indeed, a study performed by Loeken’s team [38] revealed that excessive apoptosis in the embryos of diabetic mice exhibiting NTD is accompanied by reduced expression of the Pax3 gene regulating neural tube closure in the area of the mid- and hindbrain. Subsequent experiments in embryos obtained from crosses of Pax3+/-/p53+/- males and females demonstrated that the loss of p53 function, by genetic or chemical means, prevented both apoptosis and NTDs caused by Pax-3 deficiency [77]. Based on these data, a model was proposed, in which excessive glucose metabolism inhibits the expression of Pax3 [6]. This, in turn, leads to the activation of p53-dependent apoptosis of the neuroepithelium and, consequently, to the formation of NTDs.
Our study in diabetic TNFα knockout mice revealed that the level of excessive apoptosis in the brain of TNFα-/- embryos was higher than in the brain of their TNFα-positive counterparts [23]. Recalling that TNFα may counteract diabetes-induced apoptosis, we need to ascertain the mechanism by which this preventive function of TNFα is carried out. We hypothesized that NF-κB is a target via which TNFα may positively regulate the resistance of post-implantation embryos to diabetes-induced apoptotic stimuli [23].
As mentioned above, in most cell types NF-κB exists in an inactive form in the cytoplasm but is transcriptionally active in post-implantation stage embryos. The first evidence to demonstrate the functional role of NF-κB in normal embryogenesis was obtained from studies in mice lacking the p65 subunit of NF-κB [78]. The embryos died on days 14-15 of pregnancy and this detrimental event was associated with massive hepatocyte apoptosis. Other experiments were carried out with mice lacking the inhibitory NF-κB (IκB) protein kinases 1 and 2 (IKK1 and IKK2), which are crucial for NF-κB activation [79]. The loss of NF-κB activity in these mice was associated with an increased incidence of embryos with exencephaly and excessive apoptosis in the neuronal epithelium.
The abovementioned studies suggested that NF-κB in organogenesis stage embryos acts as an apoptosis inhibitor. Teratological studies supported this suggestion. Indeed, experiments with thalidomide [80], cyclophosphamide (CP) [81] and valproic acid [82] imply that the suppression of NF-κB activity may be a mechanism by which the teratogens activate apoptosis in targeted embryonic structures. On the other hand, exposure of embryos to phenytoin resulting in non-closure of the anterior neuropore was found to be associated with NF-κB activation [83]. However, not only activation but also suppression of apoptosis may adversely affect the process of neural tube formation [84]. Therefore, the study with phenytoin does not invalidate the finding that NF-κB works as an apoptosis inhibitor in embryos.
Our study in diabetic mice revealed that malformed TNFα-/- embryos exhibit a lower level of active NF-κB complexes than TNFα+/+ embryos [23]. Because TNFα can be regarded as a powerful activator of NF-κB, we prpopose the following hypothesis: if NF-κB functions as a negative regulator of apoptosis during neural tube closure, then TNFα may act as a suppressor of diabetes-induced apoptosis by counteracting diabetes-induced suppression of NF-κB activity. Further studies are needed to investigate whether this hypothesis is correct. Nevertheless, it is worth noting that our data implicate a regulatory role on the part of NF-κB on diabetes-induced apoptotic stimuli. These data were confirmed by a recent study, in which the increased incidence of malformations was associated with a decreased NF-κB activity in embryos of STZ-induced diabetic rats [85].
Teratological studies with cyclophosphamide (CP) are also interesting in this regard. The teratogenic potential of CP is mainly associated with DNA damage induced by its metabolites such as phosphoramide mustard and acrolein [86, 87]. CP is also capable of inducing ROS and oxidative stress [88], suggesting that the teratogenic mechanism of CP may be similar to that of diabetes. Our studies implying that TNFα-mediated activation of NF-κB in embryos is a mechanism increasing their resistance to CP [89, 90] support the hypothesis for a regulatory role of TNF-α-activated NF-κB in diabetes.
The model by Loeken to explain the mechanisms of diabetes-induced NTDs suggests that accumulation of p53 is a central event in the pathway underlying diabetes-induced excessive apoptosis [6]. Our recent study revealed that CP-induced excessive apoptosis is also mediated by p53 and, importantly, that p53 mediates CP-induced suppression of NF-κB DNA binding [91]. These data suggest that Loeken’s model can legitimately be used to explain both diabetes-induced suppression of NF-κB activity and the function of TNFα as a protector against diabetes-induced teratogenic stress.
Conclusion
The data presented in this review suggest that TNFα may play a dual role in the pathogenesis of diabetes-induced embryopathies. It may act as a mediator of diabetes-induced embryotoxic stimuli leading to the death of peri-implantation stage embryos and as a suppressor of diabetes-induced apoptosis in post-implantation stage embryos. In addition, they suggest that molecules such as LIF and NF-κB may be critical players in the mechanisms determining the outcome of diabetes-induced embryopathic stress.
TNFα, LIF, NF-κB and molecules involved in NF-κB activation pathways are presently considered to be possible targets for the treatment of diabetes and diabetes-induced complications [92-94]. However, it is pointed out that therapy based on these molecules may have several detrimental consequences [95]. The problem may be aggravated, if the therapy results in modulation of a target molecule in the uterus and embryo. An increased incidence of spontaneous abortions and/or malformed offspring may be one of the unexpected side effects. Therefore, we need to learn more about the role of these molecules in the pathogenesis of diabetes-induced embryopathies.
Acknowledgments:
This work was supported by Grant 6234-1 from the Israel Ministry of Health.
References
- Platt MJ, Stanisstreet M, Casson IF, Howard CV, Walkinshaw S, Pennycook S, Mc Kendrick O. St Vincent's Declaration 10 years on: outcomes of diabetic pregnancies. Diabet Med 2002. 19:216-220. [DOD] [CrossRef]
- Mills JL, Simpson JL, Driscoll SG, Jovanovic-Peterson L, Van Allen M, Aarons JH, Metzger B, Bieber FR, Knopp RH, Holmes LB, et al. Incidence of spontaneous abortion among normal women and insulin-dependent diabetic women whose pregnancies were identified within 21 days of conception. N Engl J Med 1988. 319(25):1617-1623. [DOD]
- Greene MF, Hare JW, Cloherty JP, Benacerraf BR, Soeldner JS. First-trimester hemoglobin A1 and risk for major malformation and spontaneous abortion in diabetic pregnancy. Teratology 1989. 39(3):225-231. [DOD] [CrossRef]
- Hanson U, Persson B, Thurnell S. Relationship between haemoglobin A1c in early type 1 (insulin-dependent) diabetic pregnancy and the occurrence of spontaneous abortion and fetal malformation in Sweden. Diabetologia 1990. 33(2):100-104. [DOD] [CrossRef]
- Temple R, Aldridge V, Greenwood R, Heyburn P, Sampson M, Stanley K. Association between outcome of pregnancy and glycaemic control in early pregnancy in type 1 diabetes: population based study. BMJ 2002. 325:1275-1276. [DOD] [CrossRef]
- Loeken MR. Advances in understanding the molecular causes of diabetes-induced birth defects. J Soc Gynecol Investig 2006. 3:2-10. [DOD] [CrossRef]
- Ornoy A, Cohen AM. Teratogenic effects of sucrose diet in diabetic and nondiabetic rats. Isr J Med Sci 1980. 16:789-791. [DOD]
- Buchanan TA, Denno KM, Sipos GF, Sadler TW. Diabetic teratogenesis. In vitro evidence for a multifactorial etiology with little contribution from glucose per se. Diabetes 1994. 43:656-660. [DOD] [CrossRef]
- Eriksson UJ, Cederberg J, Wentzel P. Congenital malformations in offspring of diabetic mothers - animal and human studies. Rev Endocr Metab Disord 2003. 4:79-93. [DOD] [CrossRef]
- Torchinsky A, Toder V, Carp H, Orenstain H, Fein A. In vivo evidence for the existence of a threshold for hyperglycemia-induced major fetal malformations: relevance to the etiology of diabetic teratogenesis. Early Pregnancy 1997. 3:27-33. [DOD]
- Li R, Thorens B, Loeken MR. Expression of the gene encoding the high-Km glucose transporter 2 by the early postimplantation mouse embryo is essential for neural tube defects associated with diabetic embryopathy. Diabetologia 2007. 50:682-689. [DOD] [CrossRef]
- Loeken MR. Free radicals and birth defects. J Matern Fetal Neonatal Med 2004. 15:6-14. [DOD] [CrossRef]
- Akazawa S. Diabetic embryopathy: studies using a rat embryo culture system and an animal model. Congenit Anom (Kyoto) 2005. 45:73-79. [DOD] [CrossRef]
- Loeken MR. Current perspectives on the causes of neural tube defects resulting from diabetic pregnancy. Am J Med Genet C Semin Med Genet 2005. 135:77-87. [DOD]
- Ornoy A. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod Toxicol 2007. 24:31-41. [DOD] [CrossRef]
- Dennery P. Effects of oxidative stress on embryonic development. Birth Defects Res C Embryo Today 2007. 81:155-162. [DOD] [CrossRef]
- Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. FASEB J 2006. 20:1589-1598. [DOD] [CrossRef]
- Pampfer S. Dysregulation of the cytokine network in the uterus of the diabetic rat. Am J Reprod Immunol 2001. 45:375-381. [DOD] [CrossRef]
- Jawerbaum A, Gonzalez E. Diabetic pregnancies: the challenge of developing in a pro-inflammatory environment. Curr Med Chem 2006. 13:2127-2138. [DOD] [CrossRef]
- Torchinsky A, Toder V, Savion S, Shepshelovich J, Orenstein H, Fein A. Immunostimulation increases the resistance of mouse embryos to the teratogenic effect of diabetes mellitus. Diabetologia 1997. 40:635-640. [DOD] [CrossRef]
- Machado AF, Zimmerman EF, Hovland DN Jr, Weiss R, Collins MD. Diabetic embryopathy in C57BL/6J mice. Altered fetal sex ratio and impact of the splotch allele. Diabetes 2001. 50:1193-1199. [DOD] [CrossRef]
- Rudge MV, Damasceno DC, Volpato GT, Almeida FC, Calderon IM, Lemonica IP. Effect of Ginkgo biloba on the reproductive outcome and oxidative stress biomarkers of streptozotocin-induced diabetic rats. Braz J Med Biol Res 2007. 40(8):1095-1099. [DOD] [CrossRef]
- Torchinsky A, Gongadze M, Orenstein H, Savion S, Fein A, Toder V. TNF-alpha acts to prevent occurrence of malformed fetuses in diabetic mice. Diabetologia 2004. 47:132-139. [DOD] [CrossRef]
- Pampfer S, Wuu YD, Vanderheyden I, De Hertogh R. Expression of tumor necrosis factor-alpha (TNF-alpha) receptors and selective effect of TNFalpha on the inner cell mass in mouse blastocysts. Endocrinology 1994. 134(1):206-212. [DOD] [CrossRef]
- Pampfer S, Vanderheyden I, McCracken JE, Vesela J, De Hertogh R. Increased cell death in rat blastocysts exposed to maternal diabetes in utero and to high glucose or tumor necrosis factor-a in vitro. Development 1997. 124:4827-4836. [DOD]
- Wuu YD, Pampfer S, Becquet P, Vanderheyden I, Lee KH, De Hertogh R. Tumor necrosis factor alpha decreases the viability of mouse blastocysts in vitro and in vivo. Biol Reprod 1999. 60:479-483. [DOD] [CrossRef]
- Soto P, Natzke RP, Hansen PJ. Actions of tumor necrosis factor-alpha on oocyte maturation and embryonic development in cattle. Am J Reprod Immunol 2003. 50:380-388. [DOD] [CrossRef]
- Pampfer S, Vanderheyden I, Vesela J, De Hertogh R. Neutralization of tumor necrosis factor alpha (TNF alpha) action on cell proliferation in rat blastocysts by antisense oligodeoxyribonucleotides directed against TNF alpha p60 receptor. Biol Reprod 1995. 52:1316-1326. [DOD]
- Pampfer S. Peri-implantation embryopathy induced by maternal diabetes. J Reprod Fertil Suppl 2000. 55:129-139. [DOD]
- Kimmel CA, Price CJ. Developmental toxicity studies. In: Arnold DL, Grice HC, Krewski DR. Handbook of in vivo toxicity testing 1990. pp 271-301. [DOD]
- De Hertogh R, Vanderheyden I, Glorieux B, Ekka E. Oestrogen and progestogen receptors in endometrium and myometrium at the time of blastocyst implantation in pregnant diabetic rats. Diabetologia 1989. 32:568-572. [DOD]
- Wentzel P, Jansson L, Eriksson UJ. Diabetes in pregnancy: uterine blood flow and embryonic development in the rat. Pediatr Res 1995. 38:598-606. [DOD] [CrossRef]
- Eriksson U, Dahlström E, Larsson KS, Hellerström C. Increased incidence of congenital malformations in the offspring of diabetic rats and their prevention by maternal insulin therapy. Diabetes 1982. 31:1-6. [DOD] [CrossRef]
- Uriu-Hare JY, Stern JS, Reaven GM, Keen CL. The effect of maternal diabetes on trace element status and fetal development in the rat. Diabetes 1985. 34:1031-1040. [DOD] [CrossRef]
- Eriksson UJ, Dahlström VE, Lithell HO. Diabetes in pregnancy: influence of genetic background and maternal diabetic state on the incidence of skeletal malformations in the fetal rat. Acta Endocrinol Suppl (Copenh) 1986. 277:66-73. [DOD]
- Giavini E, Broccia ML, Prati M, Roversy GD, Vismara C. Effects of streptozotocin-induced diabetes on fetal development of the rat. Teratology 1986. 34:81-88. [DOD] [CrossRef]
- Cederberg J, Eriksson UJ. Decreased catalase activity in malformation-prone embryos of diabetic rats. Teratology 1997. 56:350-357. [DOD] [CrossRef]
- Phelan SA, Ito M, Loeken MR. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes 1997. 46:1189-1197. [DOD] [CrossRef]
- Siman CM, Eriksson UJ. Vitamin E decreases the occurrence of malformations in the offspring of diabetic rats. Diabetes 1997. 46:1054-1061. [DOD] [CrossRef]
- Novaro V, Jawerbaum A, Faletti A, Gimeno MA, Gonzalez ET. Uterine nitric oxide and prostaglandin E during embryonic implantation in non-insulin-dependent diabetic rats. Reprod Fertil Dev 1998. 10:217-223. [DOD] [CrossRef]
- Sakamaki H, Akazawa S, Ishibashi M, Izumino K, Takino H, Yamasaki H, Yamaguchi Y, Goto S, Urata Y, Kondo T, Nagataki S. Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes 1999. 48:1138-1144. [DOD] [CrossRef]
- Fein A, Kostina E, Savion S, Orenstein H, Shepshelovich J, Ornoy A, Torchinsky A, Toder V. Expression of tumor necrosis factor-alpha in the uteroplacental unit of diabetic mice: effect of maternal immunopotentiation. Am J Reprod Immunol 2001. 46:161-168. [DOD] [CrossRef]
- Fein A, Magid N, Savion S, Orenstein H, Shepshelovich J, Ornoy A, Torchinsky A, Toder V. Diabetes teratogenicity in mice is accompanied with distorted expression of TGF-beta2 in the uterus. Teratog Carcinog Mutagen 2002. 22:59-71. [DOD] [CrossRef]
- Pampfer S. Apoptosis in rodent peri-implantation embryos: differential susceptibility of inner cell mass and trophectoderm cell lineages-a review. Placenta 2000. 21(Suppl A):S3-S10. [DOD]
- Otani H, Tanaka O, Tatewaki R, Naora H, Yoneyama T. Diabetic environment and genetic predisposition as causes of congenital malformations in NOD mouse embryos. Diabetes 1991. 40:1245-1250. [DOD] [CrossRef]
- Pampfer S, Donnay I. Apoptosis at the time of embryo implantation in mouse and rat. Cell Death Differ 1999. 6:533-545. [DOD] [CrossRef]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 2001. 11(9):372-377. [DOD] [CrossRef]
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006. 25:6680-6684. [DOD] [CrossRef]
- Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NFkappaB is the answer - role of Rel/NFkappaB in the regulation of apoptosis. Oncogene 2003. 22:8961-8982. [DOD] [CrossRef]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ 2003. 10:45-65. [DOD] [CrossRef]
- Kim S, Millet I, Kim HS, Kim JY, Han MS, Lee MK, Kim KW, Sherwin RS, Karin M, Lee MS. NF-kappa B prevents beta cell death and autoimmune diabetes in NOD mice. Proc Natl Acad Sci U S A 2007. 104:1913-1918. [DOD] [CrossRef]
- Torchinsky A, Toder V. To die or not to die: the function of the transcription factor NF-kappaB in embryos exposed to stress. Am J Reprod Immunol 2004. 51:138-143. [DOD] [CrossRef]
- Nakamura H, Kimura T, Ogita K, Nakamura T, Takemura M, Shimoya K, Koyama S, Tsujie T, Koyama M, Murata Y. NF-kappaB activation at implantation window of the mouse uterus. Am J Reprod Immunol 2004. 51:16-21. [DOD] [CrossRef]
- Kimber SJ. Leukemia inhibitory factor in implantation and uterine biology. Reproduction 2005. 130:131-145. [DOD] [CrossRef]
- Torchinsky A, Markert UR, Toder V. TNF-alpha-mediated stress-induced early pregnancy loss: a possible role of leukemia inhibitory factor. Chem Immunol Allergy 2005. 89:62-71. [DOD]
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Kontgen F, Abbondanzo SJ. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 1992. 359:76-79. [DOD] [CrossRef]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 2003. 374:1-20. [DOD] [CrossRef]
- Ernst M, Inglese M, Waring P, Campbell IK, Bao S, Clay FJ, Alexander WS, Wicks IP, Tarlinton DM, Novak U, Heath JK, Dunn AR. Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation. J Exp Med 2001. 194:189-203. [DOD] [CrossRef]
- Bamberger AM, Erdmann I, Bamberger CM, Jenatschke SS, Schulte HM. Transcriptional regulation of the human 'leukemia inhibitory factor' gene: modulation by glucocorticoids and estradiol. Mol Cell Endocrinol 1997. 127:71-79. [DOD] [CrossRef]
- Laird SM, Tuckerman EM, Cork BA, Li TC. Expression of nuclear factor kappa B in human endometrium; role in the control of interleukin 6 and leukaemia inhibitory factor production. Mol Hum Reprod 2000. 6:34-40. [DOD] [CrossRef]
- Inagaki N, Stern C, McBain J, Lopata A, Kornman L, Wilkinson D. Analysis of intra-uterine cytokine concentration and matrix-metalloproteinase activity in women with recurrent failed embryo transfer. Hum Reprod 2003. 18:608-615. [DOD] [CrossRef]
- Ledee-Bataille N, Lapree-Delage G, Taupin JL, Dubanchet S, Frydman R, Chaouat G. Concentration of leukaemia inhibitory factor (LIF) in uterine flushing fluid is highly predictive of embryo implantation. Hum Reprod 2002. 17:213-218. [DOD] [CrossRef]
- Haines BP, Voyle RB, Rathjen PD. Intracellular and extracellular leukemia inhibitory factor proteins have different cellular activities that are mediated by distinct protein motifs. Mol Biol Cell 2000. 11:1369-1383. [DOD]
- Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature 2007. 450:721-724. [DOD] [CrossRef]
- Keim AL, Chi MM, Moley KH. Hyperglycemia-induced apoptotic cell death in the mouse blastocyst is dependent on expression of p53. Mol Reprod Dev 2001. 60:214-224. [DOD] [CrossRef]
- Moley KH. Hyperglycemia and apoptosis: mechanisms for congenital malformations and pregnancy loss in diabetic women. Trends Endocrinol Metab 2001. 12:78-82. [DOD] [CrossRef]
- Wyman A, Pinto A, Sheridan R, Moley KH. One-cell zygote transfer from diabetic to non-diabetic mouse results in congenital malformations and growth retardation in offspring. Endocrinology 2007. 149(2):466-469. [DOD]
- Sadler TW. Effects of maternal diabetes on early embryogenesis: II. Hyperglycemia induced exencephaly. Teratology 1980. 21:349-356. [DOD] [CrossRef]
- Hunter ES 3rd, Sadler TW. The role of the visceral yolk sac in hyperglycemia-induced embryopathies in mouse embryos in vitro. Teratology 1992. 45:195-203. [DOD] [CrossRef]
- Pinter E, Reece EA, Leranth CZ, Sanyal MK, Hobbins JC, Mahoney MJ, Naftolin F. Yolk sac failure in embryopathy due to hyperglycemia: ultrastructural analysis of yolk sac differentiation associated with embryopathy in rat conceptuses under hyperglycemic conditions. Teratology 1986. 33:73-84. [DOD] [CrossRef]
- Padmanabhan R. Etiology, pathogenesis and prevention of neural tube defects. Congenit Anom (Kyoto) 2006. 46:55-67. [DOD]
- Torchinsky A, Brokhman I, Shepshelovich J, Orenstein H, Savion S, Zaslavsky Z, Koifman M, Dierenfeld H, Fein A, Toder V. Increased TNF-alpha expression in cultured mouse embryos exposed to teratogenic concentrations of glucose. Reproduction 2003. 125:527-534. [DOD] [CrossRef]
- Sun F, Kawasaki E, Akazawa S, Hishikawa Y, Sugahara K, Kamihira S, Koji T, Eguchi K. Apoptosis and its pathway in early post-implantation embryos of diabetic rats. Diabetes Res Clin Pract 2005. 67:110-118. [DOD] [CrossRef]
- El-Bassiouni EA, Helmy MH, Abou Rawash N, El-Zoghby SM, Kamel MA, Abou Raya AN. Embryopathy in experimental diabetic gestation: assessment of PGE2 level, gene expression of cyclooxygenases and apoptosis. Br J Biomed Sci 2005. 62:161-165. [DOD]
- Gäreskog M, Cederberg J, Eriksson UJ, Wentzel P. Maternal diabetes in vivo and high glucose concentration in vitro increases apoptosis in rat embryos. Reprod Toxicol 2007. 23:63-74. [DOD] [CrossRef]
- Reece EA, Ma XD, Zhao Z, Wu YK, Dhanasekaran D. Aberrant patterns of cellular communication in diabetes-induced embryopathy in rats: II, apoptotic pathways. Am J Obstet Gynecol 2005. 192:967-972. [DOD] [CrossRef]
- Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorogenesis. Genes Dev 2002. 16:676-680. [DOD] [CrossRef]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 1995. 376:167-170. [DOD] [CrossRef]
- Li Q, Estepa G, Memet S, Israel A, Verma IM. Complete lack of NF-kappaB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev 2000. 14:1729-1733. [DOD]
- Hansen JM, Gong SG, Philbert M, Harris C. Misregulation of gene expression in the redox-sensitive NF-kappaB-dependent limb outgrowth pathway by thalidomide. Dev Dyn 2002. 225:186-194. [DOD] [CrossRef]
- Torchinsky A, Lishanski L, Wolstein O, Shepshelovich J, Orenstein H, Savion S, Zaslavsky Z, Carp H, Brill A, Dikstein R, Toder V, Fein A. NF-kappa B DNA-binding activity in embryos responding to a teratogen, cyclophosphamide. BMC Dev Biol 2002. 2:2. [DOD] [CrossRef]
- Dawson JE, Raymond AM, Winn LM. Folic acid and pantothenic acid protection against valproic acid-induced neural tube defects in CD-1 mice. Toxicol Appl Pharmacol 2006. 211:124-132. [DOD] [CrossRef]
- Kennedy JC, Memet S, Wells PG. Antisense evidence for nuclear factor-kappaB-dependent embryopathies initiated by phenytoin-enhanced oxidative stress. Mol Pharmacol 2004. 66:404-412. [DOD]
- Weil M, Jacobson MD, Raff MC. Is programmed cell death required for neural tube closure? Curr Biol 1997. 7:281-284. [DOD]
- Gäreskog M, Eriksson UJ, Wentzel P. Combined supplementation of folic acid and vitamin E diminishes diabetes-induced embryotoxicity in rats. Birth Defects Res A Clin Mol Teratol 2006. 76:483-490. [DOD] [CrossRef]
- Mirkes PE. Cyclophosphamide teratogenesis: a review. Teratog Carcinog Mutagen 1985. 5:75-88. [DOD] [CrossRef]
- Fraiser LH, Kanekal S, Kehrer JP. Cyclophosphamide toxicity. Drugs 1991. 42:781-795. [DOD]
- Hansen JM. Oxidative stress as a mechanism of teratogenesis. Birth Defects Research Part C Embryo Today 2006. 78:293-307. [DOD] [CrossRef]
- Torchinsky A, Shepshelovich J, Orenstein H, Zaslavsky Z, Savion S, Carp H, Fain A, Toder V. TNF-alpha protects embryos exposed to developmental toxicants. Am J Reprod Immunol 2003. 49:159-168. [DOD] [CrossRef]
- Torchinsky A, Gongadze M, Savion S, Fein A, Toder V. Differential teratogenic response of TNFalpha+/+ and TNFalpha-/- mice to cyclophosphamide: the possible role of NF-kappaB. Birth Defects Res A Clin Mol Teratol 2006. 76:437-444. [DOD] [CrossRef]
- Pekar O, Molotski N, Savion S, Fein A, Toder V, Torchinsky A. p53 regulates cyclophosphamide teratogenesis by controlling caspases 3, 8, 9 activation and NF-kappaB DNA binding. Reproduction 2007. 134:379-388. [DOD] [CrossRef]
- Aggarwal BB, Shishodia S, Ashikawa K, Bharti AC. The role of TNF and its family members in inflammation and cancer: lessons from gene deletion. Curr Drug Targets Inflamm Allergy 2002. 1:327-341. [DOD] [CrossRef]
- Bacher S, Schmitz ML. The NF-kappaB pathway as a potential target for autoimmune disease therapy. Curr Pharm Des 2004. 10:2827-2837. [DOD] [CrossRef]
- Skundric DS, Lisak RP. Role of neuropoietic cytokines in development and progression of diabetic polyneuropathy: from glucose metabolism to neurodegeneration. Exp Diabesity Res 2003. 4:303-312. [DOD]
- Fraser CC. Exploring the positive and negative consequences of NF-kappaB inhibition for the treatment of human disease. Cell Cycle 2006. 5:1160-1163. [DOD]
This article has been cited by other articles:
|


)


