Review
| Rev Diabet Stud,
2007,
4(4):209-225 |
DOI 10.1900/RDS.2007.4.209 |
Pleiotropic Roles of PDX-1 in the Pancreas
Hideaki Kaneto, Takeshi Miyatsuka, Dan Kawamori, Taka-aki Matsuoka
Department of Internal Medicine and Therapeutics (A8), Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan
Address correspondence to: Hideaki Kaneto, e-mail: kaneto@medone.med.osaka-u.ac.jp
Manuscript submitted January 28, 2008; resubmitted February 18, 2008; accepted February 29, 2008.
Keywords: diabetes, beta-cell differentiation, PDX-1, pancreas development, beta-cell glucose toxicity
Abstract
It is well known that pancreatic and duodenal homeobox factor-1 (PDX-1) plays a pleiotropic role in the pancreas. In the developing pancreas, PDX-1 is involved in both pancreas formation and β-cell differentiation. In mature β-cells, PDX-1 transactivates insulin and other β-cell-related genes such as GLUT2 and glucokinase. Furthermore, PDX-1 plays an important role in the induction of insulin-producing cells in various non-β-cells and is thereby a possible therapeutic target for diabetes. On the other hand, under diabetic conditions, expression and/or activity of PDX-1 in β-cells is reduced, which leads to suppression of insulin biosynthesis and secretion. It is likely that PDX-1 inactivation explains, at least in part, the molecular mechanism for β-cell glucose toxicity found in diabetes.
A variety of transcription factors are involved in pancreas formation and β-cell differentiation
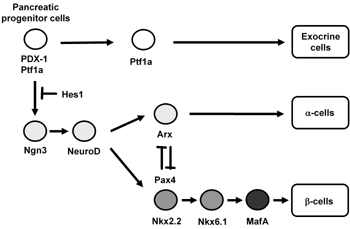
The adult pancreas is composed of exocrine (acini and ducts) and endocrine compartments (α-, β-, δ-, ε-, and PP-cells). Each of the four endocrine cell types synthesizes and secretes one hormone: glucagon (α-cells), insulin (β-cells), somatostatin (δ-cells), ghrelin (ε-cells) and pancreatic polypeptide (PP-cells). The embryonic pancreas initially develops by fusion of the dorsal and ventral buds of the primitive gut epithelium. These two types of buds grow and fuse to form the definitive pancreas [1-4]. The thickening of the dorsal and ventral surface of the foregut is observed from E8.5-E9.5 in the mouse. It has been shown that pancreatic transcription networks play a crucial role in early pancreas organogenesis and endocrine cell formation (Figure 1) [5, 6].
 |
 |
Figure 1. Pancreatic transcription factor hierarchy during pancreas development. It is well known that many transcription factors are involved in pancreas formation and β-cell differentiation. Among the various transcription factors, PDX-1 plays a crucial role in pancreas formation and β-cell differentiation as well as maintenance of mature β-cell function. Ngn3 and NeuroD are also important transcription factors for pancreatic endocrine cell differentiation. MafA expression is induced at the final stages of β-cell differentiation and functions as a potent activator of insulin gene transcription. |
|
Pancreatic and duodenal homeobox factor-1 (PDX-1) (also known as IDX-1/STF-1/IPF1) [7-9], a member of the large family of homeodomain (HD)-containing proteins, is expressed in precursors of the endocrine and exocrine compartments of the pancreas and is essential for pancreas development [10-18], β-cell differentiation [19-29], and maintenance of mature β-cell function by regulating several β-cell-related genes [30-38]. PDX-1 expression is initially observed at E8.5-E9.0 in pancreatic progenitor cells, which means that early PDX-1 expression is likely to be a useful marker of pancreatic identity. Interestingly, a study based upon temporally controlled Cre recombination demonstrated that cells expressing PDX-1 give rise to all three types of pancreatic tissue: exocrine, endocrine and duct [39, 40]. Furthermore, it was shown that exocrine and endocrine progenitors express PDX-1 throughout early embryogenesis, whereas adult duct progenitors express PDX-1 only between E9.5 and E12.5. These results suggest that the vast majority of progenitors for ducts and exocrine/endocrine cells are separated before E12.5. Another study using lineage tracing demonstrated that cells expressing another pancreatic transcription factor Ptf1a (also known as PTF1-p48) give rise to all three types of pancreatic tissue and supports the specification of precursors of all three pancreatic cell types [41]. When these two important reports are taken together, it is likely that PDX-1 and Ptf1a double-positive cells are pancreatic progenitor cells.
PDX-1 expression is maintained in precursor cells during pancreas development but becomes restricted to β-cells in the mature pancreas (Figure 1). Mice homozygous for a targeted mutation in the PDX-1 gene are apancreatic and develop fatal perinatal hyperglycemia [10], indicating that PDX-1 plays a crucial role in the formation of endocrine and exocrine cells. It is noted that PDX-1 expression is not required for pancreatic determination of the endoderm, because initial bud formation is observed in PDX-1-/- mice. Loss of PDX-1 function results in pancreatic agenesis in humans as well as in mice [15].
Differentiation into β-cells as well as maintenance of the β-cell phenotype also requires PDX-1. In mature β-cells, PDX-1 transactivates insulin and other genes involved in glucose sensing and metabolism such as GLUT2 and glucokinase [33, 34]. It was also reported that PDX-1+/- mice are glucose intolerant, with increased islet apoptosis, decreased islet mass, and abnormal islet architecture, indicating that proper gene dosage of PDX-1 is crucial for normal glucose homeostasis [16, 34, 36]. These findings are consistent with the report that humans heterozygous for an inactivating mutation of PDX-1 suffer from maturity-onset diabetes of the young (MODY 4) [42].
Furthermore, to explore the role of PDX-1 in the formation and maintenance of the pancreas, genetically engineered mice were developed using the Tet-off system, in which PDX-1 expression can be controlled by treatment of the mice with tetracycline or doxycycline [18]. In these mice, the coding region of the endogenous PDX-1 gene is replaced by a PDX-1 transgene under the control of a tetracycline-regulated transactivator (tTA). Hence, in the absence of doxycycline, tTA activates the transcription of a transgene encoding PDX-1. Expression of the transgene-encoded PDX-1 rescued the PDX-1-null phenotype, and doxycycline-mediated repression of the PDX-1 transgene throughout gestation recapitulated the PDX-1 null phenotype. Doxycycline treatment at mid pancreogenesis blocked further development of the pancreas [18]. In addition, when PDX-1 expression was shut off by doxycycline in adult mice, insulin biosynthesis was decreased and glucose homeostasis was disturbed [18]. These data further confirm the importance of PDX-1 in pancreas development, β-cell differentiation, and maintenance of mature β-cell function.
Hb9 is also a member of the large family of homeodomain (HD)-containing proteins and plays crucial roles in the early stages of pancreas development. While PDX-1 is involved in the development of the entire pancreas [5, 6, 10-18], Hb9 plays an important role in the development of the dorsal pancreas [43, 44] (Table 1). Indeed, it was shown that, in Hb9-/- mice, the dorsal pancreas is not formed and PDX-1 expression is not observed in the endodermal epithelial cells that are destined to form the dorsal pancreas. Therefore, it is likely that Hb9 functions upstream of PDX-1 in the dorsal pancreas and plays an important role in differentiation of the dorsal pancreas.
Table
1.
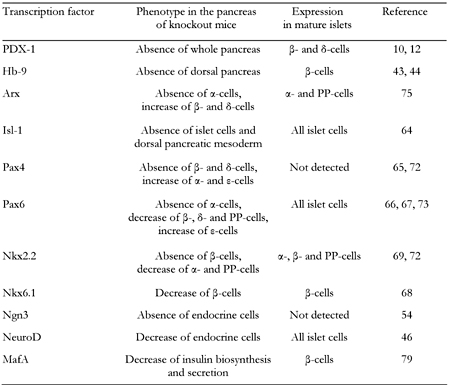
Pancreas-related phenotypes in knockout mice of pancreatic transcription factors |
|
 |
|
NeuroD and neurogenin3 (Ngn3), both of which are basic helix loop helix (bHLH) proteins, are also known to play important roles in pancreas development. NeuroD, a member of the bHLH transcription factor family, also known as BETA2, is expressed in pancreatic and intestinal endocrine cells and neural tissues. NeuroD also plays an important role in regulation of insulin gene transcription [45-48]. Mice homozygous for the null mutation in NeuroD have a striking reduction in the number of β-cells, develop severe diabetes and die perinatally [46] (Table 1). Furthermore, it has been reported that the insulin enhancer elements, E-box (NeuroD binding site) and A-box (PDX-1 binding site), are very important for insulin gene transcription [49, 50]. Neurogenin3 (Ngn3), a member of the basic helix-loop-helix (bHLH) transcription factor family, is involved in endocrine differentiation [40, 51-56]. After bud formation, Ngn3 is transiently expressed in endocrine precursor cells and functions as a potential initiator of endocrine differentiation. It has also been shown that Ngn3 directly regulates a variety of pancreatic transcription factors such as NeuroD, Pax4 and Nkx2.2 [57-60], which further strengthen the hypothesis that Ngn3 plays a crucial role in the initiation of endocrine differentiation. Transgenic mice overexpressing Ngn3 show a marked increase in endocrine cell formation, indicating that Ngn3 induces the differentiation of islet cell precursors [52, 53]. In contrast, mice with a targeted disruption in Ngn3 have no endocrine cells [54] (Table 1). Since Ngn3 is not expressed in mature β-cells, it is likely that an increase in mature β-cell numbers after birth is not due to differentiation from Ngn3-positive endocrine progenitor cells.
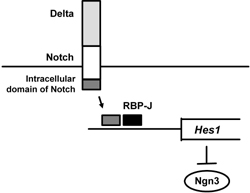
In addition, it is interesting to note that the Notch pathway plays an important role in differentiation from pancreatic progenitor cells to Ngn3-positive endocrine progenitor cells [61-63]. Indeed, it was shown that activation of the Notch pathway in pancreas progenitor cells leads to suppression of proper differentiation to both endocrine and exocrine cell lineages. After Notch activation by Delta, the intracellular domain of Notch and the mammalian Suppressor of Hairless RBP-J activate Hairy and Enhancer-of-split 1 (Hes1), which leads to suppression of Ngn3 expression (Figure 2). Therefore, it is likely that suppression of the Notch pathway leads to differentiation from pancreatic progenitor cells to Ngn3-positive endocrine cell lineage. In contrast, activation of the Notch pathway would preserve pancreatic progenitor cells from differentiation to Ngn3-positive endocrine cell lineage. Indeed, it was shown that mice deficient in Hes1 display severe pancreatic hypoplasia caused by depletion of pancreatic epithelial precursors, which is due to accelerated differentiation into endocrine cells [61]. It is probable, therefore, that Hes1 operates as a general negative regulator of endodermal endocrine differentiation.
 |
|
Figure 2. Role of the Notch pathway in pancreatic progenitor cells. After Notch activation by Delta, intracellular domain of Notch and the mammalian Suppressor of Hairless RBP-J activate Hairy and Enhancer-of-split 1 (Hes1), which leads to suppression of Ngn3 expression. Therefore, it is likely that suppression of the Notch pathway leads to differentiation from pancreatic progenitor cells to Ngn3-positive endocrine cell lineage and that, in contrast, activation of the Notch pathway preserves pancreatic progenitor cells from differentiation to Ngn3-positive endocrine cell lineage. |
|
Other subclasses of homeodomain (HD) proteins such as Arx, the LIM domain protein Isl-1, the paired domain proteins Pax4 and Pax6, and the Nkx class proteins Nkx6.1 and Nkx2.2 also play an important role in pancreas development [64-75]. The pancreas-related phenotypes observed in knockout mice of each of the homeodomain proteins are as follows (see also Table 1):
- Arx-/-, absence of α-cells and increase of β- and δ-cells [75].
- Isl-1-/-, absence of islet cells [64].
- Pax4-/-, absence of β-cells, decrease of δ-cells and increase of α- and ε-cells [65, 72].
- Pax6-/-, absence of α-cells, decrease of β-, δ- and PP-cells and increase of ε-cells [66, 67, 73].
- Nkx6.1-/-, decrease of β-cells [68].
- Nkx2.2-/-, absence of β-cells, decrease of α- and PP-cells and increase of ε-cells [68, 69, 72].
As shown in Figure 1, Pax4 and Nkx2.2 are downstream of Ngn3, and Nkx6.1 is downstream of Nkx2.2. Also, it is noted that Arx is an important transcription factor that facilitates differentiation from endocrine progenitor cells to α-cells and that Arx and Pax4 are upregulated in endocrine precursor cells of Pax4-/- and Arx-/- mice respectively [27]. Therefore, it is likely that Arx-Pax4 co-repression plays an important role in proper endocrine specification by maintaining balance between α-cell and β-cell lineages (Table 1).
Finally, MafA, a basic-leucine zipper (bLZ) transcription factor, plays an important role in the final stages of β-cell differentiation and functions as a potent transactivator for the insulin gene [76-81]. During pancreas development, MafA expression is first detected at the beginning of the principal phase of insulin-producing cell production, whereas other important transcription factors such as PDX-1 and NeuroD are expressed from the early stages of pancreas development (Figure 1). In addition, MafA is expressed only in β-cells and functions as a potent activator of insulin gene transcription, whereas PDX-1 and NeuroD are expressed in various islet cell types. It has also been reported that MafA-/- mice display glucose intolerance and develop diabetes mellitus [81]. Furthermore, in MafA-/- mice, expression of insulin 1, insulin 2, PDX-1, NeuroD and GLUT2 was decreased and glucose-, arginine-, and KCl-stimulated insulin secretion was severely impaired (Table 1).
PDX-1 plays an important role in the induction of insulin-producing cells and is a possible therapeutic target for diabetes
A decrease in the number of functioning pancreatic β-cells and insufficient insulin biosynthesis and/or secretion is the hallmark of diabetes. It is very important, therefore, to search for alternative sources to induce insulin-producing cells. For the purpose of inducing insulin-producing cells from various cells and tissues, it would be useful to mimic and reproduce the alterations in expression of various pancreatic transcription factors observed during normal pancreas development. It would also be useful to induce key pancreatic transcription factors which have the potency to induce various β-cell-related genes, including insulin, in various source cells or tissues.
It has been reported that various cells and tissues such as liver, pancreas, intestine and bone marrow can be transdifferentiated into insulin-producing cells. Furthermore, it was shown that embryonic stem cells have the potential to differentiate into insulin-producing cells [82-86], but use of these cells for the treatment of diabetes may not be appropriate from an ethical point of view. Therefore, adult tissue-derived progenitor cells have been used to induce insulin-producing cells. Pancreatic ducts, acini and non-β-cells in islets have also been shown to have the potential to differentiate into insulin-producing cells [20, 23, 24, 87-91]. In addition, since the pancreas and liver arise from adjacent regions of the endoderm in embryonic development, the liver has been thought to be a potential source for the induction of insulin-producing cells [19, 26-29, 47, 92, 93]. Intestinal epithelium-derived cells and some populations of bone marrow cells were also shown to have the potential to differentiate into insulin-producing cells [21, 22, 25, 94, 95]. In such studies, several pancreatic transcription factors were used to induce insulin-producing cells from various cells or tissues. Indeed, it was reported that adenoviral expression of PDX-1 in the liver of mice induced the expression of endogenous insulin mRNA [19].
Also, hepatic immunoreactive insulin induced by PDX-1 was processed to mature insulin which was biologically active [19]. These data indicate the capacity of PDX-1 to reprogram extrapancreatic tissues toward a β-cell phenotype, which may provide a valuable approach for generating surrogate β-cells suitable for replacing the impaired β-cell function found in diabetes. These results also demonstrate the usefulness of inducing key pancreatic transcription factors in various cells and tissues which have the potential to induce various β-cell-related genes including insulin.
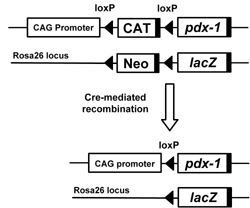
In order to carry out efficient screening of somatic tissues and cells that can transdifferentiate into β-cell-like cells in response to PDX-1, we previously generated CAG-CAT-PDX-1 mice, a transgenic line which constitutively expresses the PDX-1 gene under the control of the chicken β-actin gene (CAG) promoter after removal of the floxed stuffer sequence (CAT) by Cre-mediated recombination [26] (Figure 3). When the mice were crossed with Alb-Cre mice, which express the Cre recombinase driven by the rat albumin gene promoter, PDX-1 was expressed in hepatocytes and cholangiocytes. The PDX-1-producing liver expressed a variety of endocrine hormone genes such as insulin, glucagon, somatostatin and pancreatic polypeptide as well as exocrine genes such as elastase-1 and chymotrypsinogen 1B [26]. These mice, however, exhibited marked jaundice because of conjugated hyperbilirubinemia, and the liver tissue displayed abnormal lobe structures and multiple cystic lesions. Thus, the in vivo ectopic expression of PDX-1 in albumin-producing cells was able to initiate, although not complete, the differentiation of liver cells into insulin-producing cells. We think that this conditional PDX-1 transgenic mouse system should be useful for the efficient screening of PDX-1 responsive somatic tissues and cells (Figure 3). Given that the expression of PDX-1 continues throughout pancreas development, i.e. from the embryonic pancreatic buds to adult islets, this Cre/loxP-mediated approach would provide a suitable system for evaluating the transdifferentiation potential of PDX-1 in vivo.
 |
|
Figure 3. Tissue-specific overexpression of PDX-1 using the Cre/loxP-mediated system. We previously generated CAG-CAT-PDX-1 mice, a transgenic line which constitutively expresses the PDX-1 gene under the control of the chicken β-actin gene (CAG) promoter after the removal of the floxed stuffer sequence (CAT) by Cre-mediated recombination. When the mice were crossed with Ptf1a-Cre mice, which express the Cre recombinase driven by the Ptf1a (PTF1-p48) gene promoter, PDX-1 was expressed in precursors of all three pancreatic cell types: islets, acini, and ducts. In addition, when the mice were crossed with Alb-Cre mice, which express the Cre recombinase driven by the rat albumin gene promoter, PDX-1 was expressed in hepatocytes and cholangiocytes. |
|
In addition, it has been shown recently that a modified form of XlHbox8, the Xenopus homolog of PDX-1, carrying the VP16 transcriptional activation domain from Herpes simplex virus, efficiently induces insulin gene expression in the liver of the tadpole [96]. In this study, transgenic Xenopus tadpoles carrying the Xlhbox8-VP16 gene under the control of the transthyretin promoter were generated. Xlhbox8-VP16 was expressed only in the liver of the tadpoles. In these tadpoles, the liver was converted into a pancreas, containing both exocrine and endocrine cells. The characteristics of liver were lost from the regions converted into a pancreas [96]. In contrast, conversion of the liver to a pancreas was not observed by expression of Xlhbox8 alone (without VP16).
Following these findings in tadpoles, the effects of the PDX-1-VP16 fusion protein (PDX-1-VP16) on differentiation of cells into insulin-producing cells have been examined in mice. Indeed, it was reported recently that PDX-1-VP16 rather than wild type PDX-1 efficiently induces insulin-producing cells in the liver [27-29, 93]. In addition, it was shown that PDX-1-VP16 efficiently induces insulin gene expression in the liver, especially in the presence of the pancreatic transcription factors NeuroD or Ngn3 [27]. Although PDX-1-VP16 exerted only a slightly greater effect on the insulin promoter compared with wild type PDX-1, PDX-1-VP16, together with NeuroD or Ngn3, dramatically increased insulin promoter activity in HepG2 cells. Furthermore, when adenovirus expressing the PDX-1-VP16 fusion protein (Ad-PDX-1-VP16) was intravenously injected into mice, both insulin 1 and 2 mRNA was detected in the liver, although insulin 1 was not detected upon adenoviral induction of wild type PDX-1 (without VP-16) [27]. Ad-PDX-1-VP16 treatment, together with Ad-NeuroD or Ad-Ngn3, induced a greater insulin gene expression. After treatment with Ad-PDX-1-VP16 plus either Ad-NeuroD or Ad-Ngn3, insulin-positive cells and insulin secretory granules were observed in the liver upon immunostaining and electron microscopy, respectively [27]. Furthermore, various endocrine pancreas-related factors such as islet-type glucokinase, glucagon and somatostatin were induced after treatment with Ad-PDX-1-VP16 plus either Ad-NeuroD or Ad-Ngn3. Consequently, in STZ-induced diabetic mice, blood glucose levels were decreased by PDX-1-VP16 plus either NeuroD or Ngn3 [27].
The marked effects of PDX-1-VP16 expression, together with NeuroD or Ngn3, on insulin production and glucose tolerance indicate that this combination is useful and efficient for replacing the reduced insulin biosynthesis found in diabetes, and that PDX-1 requires the recruitment of coordinately functioning transcription factors or cofactors in order to exert its function fully. In addition, these results suggest that the synergistic activation of the insulin promoter by PDX-1 and bHLH transcription factors such as NeuroD or Ngn3 is important for the induction of insulin-producing cells from non-β-cells in order to achieve β-cell regeneration therapy in the future.
It was also shown recently that PDX-1-VP16 expressing hepatic cells were converted into functional insulin-producing cells in the presence of high glucose [28]. In this study, the authors generated a stably transfected rat hepatic cell line named WB-1 that expresses PDX-1-VP16. Expression of several genes related to endocrine pancreas development and islet function were induced by PDX-1-VP16 in the liver, although some pancreatic transcription factors were missing. In addition, these cells failed to secrete insulin upon glucose challenge. However, when WB-1 cells were transplanted into diabetic NOD-scid mice, they possessed similar properties as β-cells. Almost all β-cell-related transcription factors were induced and glucose intolerance was ameliorated [28]. In addition, in vitro culturing in high glucose medium was sufficient to induce the complete maturation of WB-1 cells into functional insulin-producing cells [28]. These results suggest that PDX-1-VP16 is very efficient and useful for replacing reduced insulin biosynthesis and for amelioration of glucose intolerance, but that PDX-1-VP16 alone is not sufficient to induce the complete transdifferentiation of various cells to functional insulin-producing cells.
Another study evaluated the effects of PDX-1-VP16 in a cell culture system as well as using hepatocytes isolated from adult rats. Adenoviral overexpression of PDX-1-VP16 efficiently converted hepatocytes into insulin-producing cells. In addition, immunoreactivity of albumin was downregulated in the transdifferentiated cells and some cells lost albumin expression almost completely [93]. These results add further weight to the hypothesis that hepatocytes possess the potential to transdifferentiate into insulin-producing cells.
Many studies have been performed to overexpress pancreatic transcription factors in different tissues using various virus-mediated approaches, but such approaches would be difficult to apply in clinical medicine. Therefore, new strategies are necessary to deliver safely various pancreatic transcription factors. Protein transduction domains (PTDs) such as the small PTD from the TAT protein of human immunodeficiency virus-1 (HIV-1), the VP22 protein of Herpes simplex virus and the third α-helix of the homeodomain of Antennapedia, a Drosophila transcription factor, are known to allow various proteins and peptides to be efficiently delivered into cells through the plasma membrane. For this reason, there has been increasing interest in their potential usefulness for the delivery of bioactive proteins and peptides into cells [23, 48, 56]. With regard to the potential of pancreatic transcription factors as therapeutic targets, the protein delivery system appears to be very promising at this point, because of the practical difficulties in applying virus-mediated approaches to clinical medicine without side effects.
In order to induce surrogate β-cells and apply them to clinical medicine, it would be advantageous to deliver key pancreatic transcription factors into pancreatic source cells and tissues using the protein delivery system. It was shown recently that the PDX-1 protein can enter various cells on its own because of an Antennapedia-like protein transduction domain sequence in its structure and that transduced PDX-1 functions similarly to endogenous PDX-1: it binds to the insulin promoter and activates its expression [23]. In addition, it was shown that PDX-1 protein transduction occurs by endocytosis and its subsequent release from endosome, followed by homogenous location in the cytoplasm and nuclei [97]. More recently, it was shown that the NeuroD protein can also enter various cells on its own because of an arginine- and lysine-rich protein transduction domain in its structure and that transduced NeuroD functions similarly to endogenous NeuroD [48]. These data clearly suggest that PDX-1 and NeuroD transduction would be a safe and valuable strategy for inducing surrogate β-cells from non-β-cells without requiring gene transfer technology.
A variety of pancreatic transcription factors are involved in PDX-1 gene expression
Since PDX-1 plays a crucial role in pancreas development, β-cell differentiation and maintenance of mature β-cell function, it is very important to understand the regulation of PDX-1 expression in the pancreas. It has been reported that PDX-1 activity is regulated by various nutrients such as glucose and insulin. It was shown that a high concentration of glucose and/or insulin increased PDX-1 DNA binding activity to the insulin gene promoter region through activation of phosphatidylinositol 3-kinase (PI3-kinase) and p38 mitogen-activated protein kinase (MAPK) [98-102]. In addition, PDX-1 gene transcription is regulated by various pancreatic transcription factors (Figure 4).
 |
|
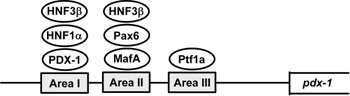
Figure 4. A variety of pancreatic transcription factors are involved in PDX-1 gene expression. Alignment of the mouse and human PDX-1 gene sequences revealed three conserved regions (Area I, II and III) which harbor binding sites of various transcription factors such as HNF-3β, HNF-1α, PDX-1, Pax6, MafA and Ptf1a. |
|
Normal endocrine pancreas development and function depend on a highly integrated transcription factor network, and subtle abnormalities in islets caused by heterozygosity or reduced gene dosage of MODY susceptibility genes lead to diabetes in humans [103]. Promoter analyses of genes involved in β-cell differentiation and function suggest complex genetic interactions among these factors. Indeed, alignment of the mouse and human PDX-1 gene sequences revealed three conserved regions referred to collectively as Area I-II-III. The Area I-II-III region harbors binding sites for MODY transcription factors such as HNF-1α (Foxa1) and PDX-1 itself as well as other transcriptional regulators such as HNF-3β (Foxa2), Pax6, MafA and HNF-6 (OC-1) [104-113], and it has been shown that PDX-1 gene transcription is actually regulated by these various pancreatic transcription factors.
It has recently been reported that another pancreatic transcription factor, Ptf1a (also known as PTF1-p48), regulates PDX-1 gene expression (Figure 4) [104]. Ptf1a, a member of the basic helix-loop-helix (bHLH) family, is known to be expressed in pancreatic progenitor cells and to bind to the mammalian Suppressor of Hairless (RBP-J) within the PTF1 complex [115, 116]. In addition, all three factors (PDX-1, Ptf1a and RBP-J) have been shown to be essential for early pancreas development [41, 117-119]. Reporter gene analyses showed that Ptf1a transactivates the PDX-1 gene promoter in pancreatic Panc-1 cells, which is enhanced by RBP-J. The Ptf1a binding site was also identified in the well-conserved regulatory sequence domain termed Area III. In addition, adenoviral overexpression of Ptf1a, together with RBP-J, markedly increased PDX-1 expression levels in pancreatic AR42J-B13 cells, which have been reported to differentiate into insulin-producing cells [120, 121]. Furthermore, it was recently demonstrated using Cre-mediated lineage tracing in mice that Area III mediates pancreas-wide PDX-1 expression during early pancreas development and that Ptf1a occupies sequences within Area III in pancreatic buds [122].
These results strongly suggest a novel transcriptional network in which Ptf1a regulates PDX-1 gene expression through binding to Area III in pancreatic progenitor cells. It is noted, however, that since PDX-1 expression is likely to be regulated in a different manner at each stage of pancreas development, in vitro promoter analysis does not necessarily recapitulate the regulation of PDX-1 expression in the developing pancreas. Therefore, the study using mice with deletion of a specific PDX-1 promoter region is very useful in evaluating the regulation of PDX-1 expression in the developing pancreas.
It has been reported that islet-specific and β-cell-specific cis-regulatory regions overlap with Area I-II-III, suggesting that Area I-II-III functions specifically in the differentiation and maintenance of pancreatic islets [104-113]. It has also recently been reported that deletion of Area I-II-III from the endogenous PDX-1 locus results in a decreased level as well as abnormal spatiotemporal expression of the PDX-1 protein. In addition, the pancreas of homozygous Area I-II-III knockout mice did not undergo ventral pancreatic bud specification and demonstrated early-onset hypoplasia in the dorsal bud [123]. In these mice, acinar tissue formed in the hypoplastic dorsal bud, but endocrine maturation was greatly impaired. In addition, while the pylorus was distorted and Brunner's glands were not observed in PDX-1-/- mice, these structures formed normally in the homozygous Area I-II-III deletion mutant mice. These results suggest that Area I-II-III is not essential for extra-pancreatic expression of PDX-1. Furthermore, Area I-II-III heterozygous knockout mice had abnormal islets and showed more severe glucose intolerance compared to PDX-1+/- mice [123]. These results supply further confirmation of the importance of Area I-II-III in pancreas formation and maintenance of β-cell function.
While PDX-1 is expressed in pancreatic progenitor cells and plays a crucial role in pancreas development and β-cell differentiation, PDX-1 expression is downregulated in exocrine and ductal cells after late embryonic development. On the other hand, re-upregulation of PDX-1 has been reported in human patients and several mouse models with pancreatic cancer and pancreatitis [124-126]. We have recently reported that programmed downregulation of PDX-1 is required for exocrine tissue formation during pancreas differentiation and that persistent expression of PDX-1 causes acinar-to-ductal metaplasia [127]. To determine whether the sustained expression of PDX-1 affects pancreas development, PDX-1 was constitutively expressed in all pancreatic lineages by transgenic approaches.
As mentioned earlier, we previously generated CAG-CAT-PDX-1 mice, a transgenic line which constitutively expresses the PDX-1 gene under the control of the chicken β-actin gene (CAG) promoter after the removal of the floxed stuffer sequence (CAT) by Cre-mediated recombination [26] (Figure 3). When these mice were crossed with Ptf1a-Cre mice, which express the Cre recombinase driven by the Ptf1a (PTF1-p48) gene promoter [41], PDX-1 was expressed in precursors of all three pancreatic cell types: islets, acini, and ducts. Two weeks after birth, the whole pancreas of the Ptf1a-Cre, CAG-CAT-PDX-1 mouse was much smaller compared to the non-transgenic pancreas, and marked abnormalities in the exocrine tissue were observed. While acinar areas with normal morphology substantially disappeared in the transgenic pancreas, a large number of cells with duct-like morphology were observed [127]. Severe atrophic cells and abnormal duct-like morphology were observed exclusively in the cells expressing exogenous PDX-1, suggesting that the phenotypes in the transgenic pancreas are caused by the cell-autonomous effect of PDX-1.
To induce exogenous expression of PDX-1 selectively in the exocrine lineage, CAG-CAT-PDX-1 mice were crossed with the resulting transgenic Elastase-Cre mice, after which recombination occurred primarily in the exocrine lineage [128]. Furthermore, lineage tracing was performed using Ptf1a-Cre, CAG-CAT-PDX1, ROSA26-lacZ and Elastase-Cre, CAG-CAT-PDX1, ROSA26-lacZ mice (Figure 3). Interestingly, a large number of duct-like cells, marked as blue β-galactosidase-positive cells, were observed in the pancreas of Elastase-Cre, CAG-CAT-PDX1 and ROSA26-lacZ mice, similar to those seen in the pancreas of Ptf1a-Cre, CAG-CAT-PDX1 and ROSA26-lacZ mice [127]. In addition, in immunostaining for BrdU and Ki67, cell proliferation was not observed in these duct-like cells.
When these results are considered together, we think that duct-like cells were induced by acinar-to-ductal transdifferentiation rather than by self-proliferation of duct cells. In summary, it is likely that programmed downregulation of PDX-1 is required for exocrine formation and that persistent upregulation of PDX-1 is sufficient to induce acinar-to-ductal metaplasia in the exocrine lineage.
Expression and/or activity of PDX-1 in β-cells are reduced under diabetic conditions and are likely to be involved in pancreatic β-cell glucose toxicity
Under diabetic conditions, chronic hyperglycemia causes the gradual deterioration of pancreatic β-cell function. This process is often observed in diabetic subjects and is clinically well known as β-cell glucose toxicity [129-133]. It has been shown that in the diabetic state, hyperglycemia per se and subsequent production of oxidative stress decrease insulin gene expression and secretion [129-145]. It has also been shown that the loss of insulin gene expression is accompanied by decreased expression and/or DNA binding activity of PDX-1 [129, 130, 137-139]. After chronic exposure to a high glucose concentration, PDX-1 expression and/or its DNA binding activity are reduced. Abnormalities in lipid metabolism have also been proposed as contributing factors to the deterioration in pancreatic β-cell function. Prolonged exposure to excessive concentrations of fatty acids inhibits insulin gene expression and secretion [146-148]. Furthermore, it has been shown recently that prolonged exposure of islets to palmitate inhibits insulin gene transcription by impairing the nuclear localization of PDX-1 [149].
Under diabetic conditions, hyperglycemia induces oxidative stress, which is involved in the β-cell glucose toxicity found in diabetes, through various pathways such as the electron transport chain in mitochondria, the non-enzymatic glycosylation reaction and the NADPH oxidase pathway [136-145, 150-153]. β-cells express GLUT2, a high-Km glucose transporter, and thereby display highly efficient glucose uptake when exposed to a high glucose concentration. In addition, β-cells are rather vulnerable to oxidative stress because of the relatively low expression of antioxidant enzymes such as catalase and glutathione peroxidase [154, 155]. Indeed, it was shown that expression of the oxidative stress markers 8-hydroxy-2'-deoxyguanosine (8-OHdG) and 4-hydroxy-2, 3-nonenal (4-HNE) were increased in islets under diabetic conditions [136, 143]. It has also been shown that when β-cell-derived cell lines or rat isolated islets are exposed to oxidative stress, insulin gene promoter activity and mRNA expression are suppressed [137-139, 141-144]. When those cells or rat isolated islets were exposed to oxidative stress, binding of PDX-1 to the insulin gene promoter was markedly reduced. Furthermore, it was shown that the decrease of insulin gene expression after chronic exposure to a high glucose concentration could be prevented by treatment with antioxidants [138, 139, 142-144]. Reduction of the expression and/or DNA binding activity of PDX-1 by chronic exposure to a high glucose concentration was also prevented by antioxidant treatment.
These results suggest that chronic hyperglycemia suppresses insulin biosynthesis and secretion by provoking oxidative stress, accompanied by the reduction of PDX-1 expression and/or its DNA binding activity. Therefore, it is likely that PDX-1 inactivation explains, at least in part, the suppression of insulin biosynthesis and secretion and is thus involved in β-cell glucose toxicity (Figure 5).
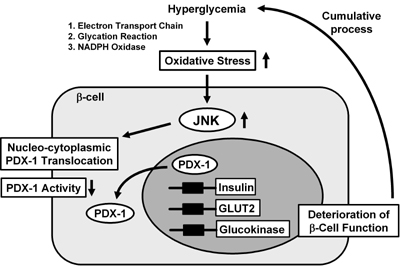 |
|
Figure 5. Nucleo-cytoplasmic translocation of PDX-1 is induced by oxidative stress and the subsequent activation of the JNK pathway. Oxidative stress and the subsequent activation of the JNK pathway induce the nucleo-cytoplasmic translocation of PDX-1, which leads to reduction of its DNA binding activity and suppression of insulin biosynthesis. It is likely that oxidative stress and the JNK pathway are involved in the β-cell dysfunction found in type 2 diabetes. |
|
In order to evaluate the role of oxidative stress in diabetes in vivo, obese diabetic C57BL/KsJ-db/db mice were treated with antioxidants (N-acetyl-L-cysteine plus vitamin C and E) [138]. Antioxidant treatment did not affect glucose-stimulated insulin secretion and moderately ameliorated glucose tolerance. β-cell mass was significantly larger in mice treated with the antioxidants. Insulin content and insulin mRNA levels were also preserved by the antioxidant treatment. Furthermore, PDX-1 expression was more clearly visible in the nuclei of islet cells after the antioxidant treatment [138]. Similar effects were observed using Zucker diabetic fatty rats, another model animal for type 2 diabetes [139]. Taken together, these data indicate that antioxidant treatment can protect β-cells against glucose toxicity.
In addition, we examined the possible anti-diabetic effects of probucol, an antioxidant widely used as an anti-hyperlipidemic agent, on the preservation of β-cell function in diabetic C57BL/KsJ-db/db mice [143]. Immunostaining for oxidative stress markers such as 4-hydroxy-2-nonenal (HNE)-modified proteins and heme oxygenase-1 revealed that probucol treatment decreases ROS in β-cells of diabetic mice. Probucol treatment also preserved β-cell mass, insulin content and glucose-stimulated insulin secretion, leading to improvement of glucose tolerance [143]. These data suggest the potential usefulness of antioxidants for diabetes and provide further support for the involvement of oxidative stress in the β-cell glucose toxicity found in diabetes.
It has been suggested that activation of the c-Jun N-terminal kinase (JNK) pathway is involved in the pancreatic β-cell dysfunction found in diabetes. It was reported that activation of the JNK pathway is involved in reduction of insulin gene expression by oxidative stress and that suppression of the JNK pathway can protect β-cells from oxidative stress [156]. When isolated rat islets were exposed to oxidative stress, the JNK pathway was activated, preceding the decrease of insulin gene expression. Adenoviral overexpression of a dominant-negative type JNK1 (DN-JNK) inhibited the decrease in insulin gene expression and secretion resulting from oxidative stress. Moreover, overexpression of wild type JNK1 (WT-JNK) suppressed both insulin gene expression and secretion [156]. These results were correlated with the reduction of PDX-1 binding to the insulin promoter. Adenoviral overexpression of DN-JNK preserved PDX-1 DNA binding activity in the presence of oxidative stress, while WT-JNK overexpression decreased PDX-1 DNA binding activity [156]. Thus, JNK-mediated suppression of PDX-1 DNA binding activity probably accounts for some of the suppression of insulin gene transcription upon oxidative stress.
In summary, it is likely that activation of the JNK pathway leads to decreased PDX-1 activity and the consequent suppression of insulin gene transcription found in the diabetic state (Figure 5). Furthermore, as a potential mechanism for JNK-mediated PDX-1 inactivation, it was recently reported that PDX-1 translocates from the nucleus to the cytoplasm in response to oxidative stress. When β-cell-derived HIT cells were subjected to oxidative stress, PDX-1 translocates from the nucleus to the cytoplasm [157]. Addition of DN-JNK inhibited this translocation, suggesting an essential role of the JNK pathway in mediating this phenomenon. In addition, leptomycin B, a specific inhibitor of the classical, leucine-rich nuclear export signal (NES), inhibited the nucleo-cytoplasmic translocation of PDX-1 induced by oxidative stress. Indeed, we identified an NES at position 82-94 of the mouse PDX-1 protein [157]. Conclusively, it is likely that oxidative stress induces the nucleo-cytoplasmic translocation of PDX-1 through activation of the JNK pathway, which leads to reduction of its DNA binding activity and suppression of insulin biosynthesis (Figure 5).
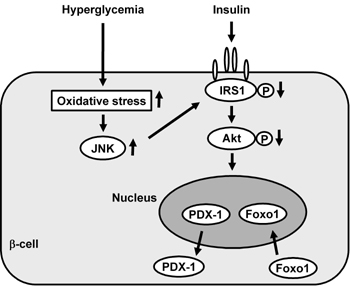
Furthermore, while the role of the forkhead transcription factor Foxo1 in β-cell function has attracted considerable attention [158, 159], we have recently reported that Foxo1 plays a role as a mediator between the JNK pathway and PDX-1 [160]. In β-cell-derived HIT cells, the intracellular localization of Foxo1 changed from the cytoplasm to the nucleus under oxidative stress conditions. In contrast to Foxo1, as mentioned above, the amount of nuclear PDX-1 decreased and its cytoplasmic distribution was increased by oxidative stress. JNK overexpression also induced the nuclear localization of Foxo1, although, on the other hand, suppression of the JNK pathway reduced the oxidative stress-induced nuclear localization of Foxo1, suggesting the involvement of the JNK pathway in Foxo1 translocation [160]. In addition, oxidative stress or activation of the JNK pathway decreased the activity of Akt in HIT cells, leading to decreased phosphorylation of Foxo1 following nuclear localization. Furthermore, adenoviral Foxo1 overexpression reduced the nuclear expression of PDX-1, whereas repression of Foxo1 by a Foxo1-specific small interfering RNA resulted in retained nuclear expression of PDX-1 under oxidative stress conditions [160]. When considered as a whole, these data indicate that oxidative stress and the subsequent activation of the JNK pathway induce nuclear translocation of Foxo1 through the modification of insulin signaling in β-cells, which leads to the nucleo-cytoplasmic translocation of PDX-1 and reduction of its DNA binding activity (Figure 6). Finally, we think that suppression of oxidative stress and/or inactivation of the JNK pathway protects β-cells from glucose toxicity found in diabetes and thus are potential therapeutic targets for diabetes.
 |
|
Figure 6. Involvement of Foxo1 in the nucleo-cytoplasmic translocation of PDX-1 which is induced by oxidative stress and the subsequent activation of the JNK pathway. Oxidative stress and the subsequent activation of the JNK pathway induce nuclear translocation of Foxo1 through modification of insulin signaling in β-cells, which leads to the nucleo-cytoplasmic translocation of PDX-1 and reduction of its DNA binding activity. |
|
Conclusions
The number of diabetic patients is dramatically increasing all over the world, and diabetes has recently been recognized as one of the most prevalent and serious metabolic diseases. Although pancreas and islet transplantation have achieved beneficial effects for type 1 diabetic patients, the availability of insulin-producing cells is limited and life-time immunosuppressive therapy is required. It is very important, therefore, to search for alternative sources to induce insulin-producing cells.
PDX-1 is a pancreatic transcription factor which plays a crucial role in pancreas formation, β-cell differentiation and maintenance of mature β-cell function. Furthermore, it is likely that PDX-1 plays a crucial role in inducing insulin-producing cells in various non-β-cells and thus could be a therapeutic target for type 1 diabetes. It is noted, however, that current strategies involve some problems with the differentiation of various cells into insulin-producing cells. For example, although insulin biosynthesis and secretion can be induced in several types of non-β-cells, it is very difficult to obtain substantial glucose-responsive insulin secretion, which is very important to maintain normal glucose tolerance.
Under diabetic conditions, chronic hyperglycemia gradually leads to the deterioration of β-cell function, which is often observed in type 2 diabetic subjects and is clinically well known as β-cell glucose toxicity. These phenomena are accompanied by a reduction in the expression and activity of pancreatic transcription factors. Therefore, it is likely that PDX-1 plays an important role in mediating mature β-cell function and that PDX-1 inactivation is involved in the β-cell glucose toxicity found type 2 diabetes.
Acknowledgments:
We thank Dr. Helena Akiko Popiel (Osaka University Graduate School of Medicine) for valuable comments on the manuscript.
References
- Slack JM. Developmental biology of the pancreas. Development 1995. 121:1569-1580. [DOD]
- Edlund H. Transcribing pancreas. Diabetes 1998. 47:1817-1823. [DOD] [CrossRef]
- Edlund H. Pancreatic organogenesis-developmental mechanisms and implications for therapy. Nat Rev Genet 2002. 3:524-532. [DOD] [CrossRef]
- Murtaugh LC. Pancreas and beta-cell development: from the actual to the possible. Development 2007. 134:427-438. [DOD] [CrossRef]
- Melloul D. Transcription factors in islet development and physiology: role of PDX-1 in beta-cell function. Ann N Y Acad Sci 2004. 1014:28-37. [DOD] [CrossRef]
- Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia 2004. 47:597-613. [DOD] [CrossRef]
- Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing-transactivator of the insulin gene. EMBO J 1993. 12:4251-4259. [DOD]
- Miller CP, McGehee RE, Habener JF. IDX-1: a new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J 1994. 13:1145-1156. [DOD]
- Leonard J, Peers B, Johnson T, Ferreri K, Lee S, Montminy MR. Characterization of somatostatin transactivating factor-1, a novel homeobox factor that stimulates somatostatin expression in pancreatic islet cells. Mol Endocrinol 1993. 7:1275-1283. [DOD] [CrossRef]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1993. 37:606-609. [DOD]
- Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, Teitelmen G. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development 1995. 121:11-18. [DOD]
- Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 1996. 122:1409-1416. [DOD]
- Offield MF, Jetton TL, Labosky P, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreas outgrowth and differentiation of the rostral duodenum. Development 1996. 122:983-985. [DOD]
- Kaneto H, Miyagawa J, Kajimoto Y, Yamamoto K, Watada H, Umayahara Y, Hanafusa Y, Matsuzawa Y, Yamasaki Y, Higashiyama S, Taniguchi N. Expression of heparin-binding epidermal growth factor-like growth factor during pancreas development: a potential role of PDX-1 in transcriptional activation. J Biol Chem 1997. 272:29137-29143. [DOD] [CrossRef]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 1997. 15:106-110. [DOD] [CrossRef]
- Dutta S, Bonner-Weir S, Montminy M, Wright C. Regulatory factor linked to late-onset diabetes? Nature 1998. 392:560. [DOD]
- Stoffers DA, Heller RS, Miller CP, Habener JF. Developmental expression of the homeodomain protein IDX-1 in mice transgenic for an IDX-1 promoter/LacZ transcriptional reporter. Endocrinology 1999. 140:5374-5381. [DOD] [CrossRef]
- Holland AM, Hale MA, Kagami H, Hammer RE, MacDonald RJ. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci USA 2002. 99:12236-12241. [DOD] [CrossRef]
- Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I, Barshack I, Seijffers R, Kopolovic J, Kaiser N, Karasik A. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med 2000. 6:568-572. [DOD] [CrossRef]
- Heller RS, Stoffers DA, Bock T, Svenstrup K, Jensen J, Horn T, Miller CP, Habener JF, Madsen OD, Serup P. Improved glucose tolerance and acinar dysmorphogenesis by targeted expression of transcription factor PDX-1 to the exocrine pancreas. Diabetes 2001. 50:1553-1561. [DOD] [CrossRef]
- Kojima H, Nakamura T, Fujita Y, Kishi A, Fujimiya M, Yamada S, Kudo M, Nishio Y, Maegawa H, Haneda M, Yasuda H, Kojima I, Seno M, Wong NC, Kikkawa R, Kashiwagi A. Combined expression of pancreatic duodenal homeobox 1 and islet factor 1 induces immature enterocytes to produce insulin. Diabetes 2002. 51:1398-1408. [DOD] [CrossRef]
- Yoshida S, Kajimoto Y, Yasuda T, Watada H, Fujitani Y, Kosaka H, Gotow T, Miyatsuka T, Umayahara Y, Yamasaki Y, Hori M. PDX-1 induces differentiation of intestinal epithelioid IEC-6 into insulin-producing cells. Diabetes 2002. 51:2505-2513. [DOD] [CrossRef]
- Noguchi H, Kaneto H, Weir GC, Bonner-Weir S. PDX-1 protein containing its own Antennapedia-like protein transduction domain can transduce pancreatic duct and islet cells. Diabetes 2003. 52:1732-1737. [DOD] [CrossRef]
- Taniguchi H, Yamato E, Tashiro F, Ikegami H, Ogihara T, Miyazaki J. Beta-cell neogenesis induced by adenovirus-mediated gene delivery of transcription factor pdx-1 into mouse pancreas. Gene Ther 2003. 10:15-23. [DOD] [CrossRef]
- Tang DQ, Cao LZ, Burkhardt BR, Xia CQ, Litherland SA, Atkinson MA, Yang LJ. In vivo and in vitro characterization of insulin-producing cells obtained from murine bone marrow. Diabetes 2004. 53:1721-1732. [DOD] [CrossRef]
- Miyatsuka T, Kaneto H, Kajimoto Y, Hirota S, Arakawa Y, Fujitani Y, Umayahara Y, Watada H, Yamasaki Y, Magnuson MA, Miyazaki J, Hori M. Ectopically expressed PDX-1 in liver initiates endocrine and exocrine pancreas differentiation but causes dysmorphogenesis. Biochem Biophys Res Commun 2003. 310:1017-1025. [DOD] [CrossRef]
- Kaneto H, Nakatani Y, Miyatsuka T, Matsuoka T, Matsuhisa M, Hori M, Yamasaki Y. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes 2005. 54:1009-1022. [DOD] [CrossRef]
- Cao LZ, Tang DQ, Horb ME, Li SW, Yang LJ. High glucose is necessary for complete maturation of Pdx 1-VP16-expressing hepatic cells into functional insulin-producing cells. Diabetes 2004. 53:3168-3178. [DOD] [CrossRef]
- Imai J, Katagiri H, Yamada T, Ishigaki Y, Ogihara T, Uno K, Hasegawa Y, Gao J, Ishihara H, Sasano H, Mizuguchi H, Asano T, Oka Y. Constitutively active PDX1 induced efficient insulin production in adult murine liver. Biochem Biophys Res Commun 2005. 326:402-409. [DOD] [CrossRef]
- Petersen HV, Serup P, Leonard J, Michelsen BK, Madsen OD. Transcriptional regulation of the human insulin gene is dependent on the homeodomain protein STF1/IPF1 acting through the CT boxes. Proc Natl Acad Sci USA 1994. 91:10465-10469. [DOD] [CrossRef]
- Peers B, Leonard J, Sharma S, Teitelman G, Montminy MR. Insulin expression in pancreatic islet cells relies on cooperative interactions between the helix loop helix factor E47 and the homeobox factor STF-1. Mol Endocrinol 1994. 8:1798-1806. [DOD] [CrossRef]
- Waeber G, Thompson N, Nicod P, Bonny C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol Endocrinol 1996. 10:1327-1334. [DOD] [CrossRef]
- Watada H, Kajimoto Y, Umayahara Y, Matsuoka T, Kaneto H, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y. The human glucokinase gene beta-cell-type promoter: An essential role of insulin promoter factor 1 (IPF1)/PDX-1 in its activation in HIT-T15 cells. Diabetes 1996. 45:1478-1488. [DOD] [CrossRef]
- Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev 1998. 12:1763-1768. [DOD] [CrossRef]
- Wang H, Maechler P, Ritz-Laser B, Hagenfeldt KA, Ishihara H, Philippe J, Wollheim CB. Pdx1 level defines pancreatic gene expression pattern and cell lineage differentiation. J Biol Chem 2001. 276:25279-25286. [DOD] [CrossRef]
- Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, Wright CV, Powers AC. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem 2002. 277:1125-11232. [DOD] [CrossRef]
- Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest 2004. 114:828-836. [DOD]
- Holland AM, Gonez LJ, Naselli G, MacDonald RJ, Harrison LC. Conditional expression demonstrates the role of the homeodomain transcription factor Pdx1 in maintenance and regeneration of beta-cells in the adult pancreas. Diabetes 2005. 54:2586-2595. [DOD] [CrossRef]
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000. 127:2317-2322. [DOD]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and distinct from duct progenitors. Development 2002. 129:2447-2457. [DOD]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet 2002. 32:128-134. [DOD] [CrossRef]
- Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 1997. 17(2):138-139. [DOD] [CrossRef]
- Harrison KA, Thaler J, Pfaff SL, Gu H, Kehrl JH. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat Genet 1999. 23:71-75. [DOD]
- Li H, Arber S, Jessell TM, Edlund H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet 1999. 23:67-70. [DOD]
- Naya FJ, Stellrecht CM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev 1995. 9:1009-1019. [DOD] [CrossRef]
- Naya FJ, Huang H, Qiu Y, Mutoh H, DeMayo F, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev 1997. 11:323-2334. [DOD] [CrossRef]
- Kojima H, Fujimiya M, Matsumura K, Younan P, Imaeda H, Maeda M, Chan L. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med 2003. 9:595-603. [DOD] [CrossRef]
- Noguchi H, Bonner-Weir S, Wei FY, Matsushita M, Matsumoto S. BETA2/NeuroD protein can be transduced into cells due to an arginine- and lysine-rich sequence. Diabetes 2005. 54:2859-2866. [DOD] [CrossRef]
- Sharma A, Stein R. Glucose-induced transcription of the insulin gene is mediated by factors required for beta-cell-type-specific expression. Mol Cell Biol 1994. 14:871-879. [DOD]
- German MS, Wang J. The insulin gene contains multiple transcriptional elements that respond to glucose. Mol Cell Biol 1994. 14:4067-4075. [DOD]
- Grapin-Botton A, Majithia AR, Melton DA. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev 2001. 15:444-454. [DOD] [CrossRef]
- Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, de Angelis MH, Lendahl U, Edlund H. Notch signaling controls pancreatic cell differentiation. Nature 1999. 400:877-881. [DOD] [CrossRef]
- Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 2000. 127:3533-3542. [DOD]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 2000. 97(4):1607-1611. [DOD] [CrossRef]
- Watada H. Neurogenin 3 is a key transcription factor for differentiation of the endocrine pancreas. Endocr J 2004. 51:255-264. [DOD] [CrossRef]
- Dominguez-Bendala J, Klein D, Ribeiro M, Ricordi C, Inverardi L, Pastori R, Edlund H. TAT-mediated neurogenin 3 protein transduction stimulates pancreatic endocrine differentiation in vitro. Diabetes 2005. 54:720-726. [DOD] [CrossRef]
- Huang HP, Liu M, Heithem M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol 2000. 20:3292-3307. [DOD] [CrossRef]
- Watada H, Scheel DW, Leung J, German MS. Distinct gene expression programs function in progenitor and mature islet cells. J Biol Chem 2003. 278:17130-17140. [DOD] [CrossRef]
- Smith SB, Gasa R, Watada H, Wang J, Griffen SC, German MS. Neurogenin3 and hepatic nuclear factor 1 cooperate in activating pancreatic expression of Pax4. J Biol Chem 2003. 278:38254-38259. [DOD] [CrossRef]
- Watada H, Mirmira RG, Leung J, German MS. Transcriptional and translational regulation of beta-cell differentiation factor Nkx6.1. J Biol Chem 2000. 275:34224-34230. [DOD]
- Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by Hes-1. Nat Genet 2000. 25:36-44. [DOD]
- Sumazaki R, Shiojiri N, Isoyama S, Masu M, Keino-Masu K, Osawa M, Nakauchi H, Kageyama R, Matsui A. Conversion of biliary system to pancreatic tissue in Hes1-deficient mice. Nat Genet 2004. 36:83-87. [DOD] [CrossRef]
- Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci USA 2003. 100:14920-14925. [DOD] [CrossRef]
- Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature 1997. 385:257-260. [DOD] [CrossRef]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 1997. 386:399-402. [DOD] [CrossRef]
- St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature 1997. 387(6631):406-409. [DOD] [CrossRef]
- Sander M, Neubuser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev 1997. 11:1662-1673. [DOD] [CrossRef]
- Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, German M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 2000. 127:5533-5540. [DOD]
- Sussel L, Kalamaras J, Hartigan-O'Connor DJ, Meneses JJ, Pedersen RA, Rubenstein JL, German MS. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development 1998. 125:2213-2221. [DOD]
- Fujitani Y, Kajimoto Y, Yasuda T, Matsuoka T, Kaneto H, Umayahara Y, Fujita N, Watada H, Miyazaki J, Yamasaki Y, Hori M. Identification of a portable repression domain and an E1A-responsive activation domain in Pax 4: a possible role of Pax 4 as a transcriptional repressor in the pancreas. Mol Cell Biol 1999. 19:8281-8291. [DOD]
- Smith SB, Ee HC, Conners JR, German MS. Paired-homeodomain transcription factor PAX4 acts as a transcriptional repressor in early pancreatic development. Mol Cell Biol 1999. 19:8272-8280. [DOD]
- Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B, Sussel L. Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proc Natl Acad Sci USA 2004. 101:2924-2929. [DOD] [CrossRef]
- Heller RS, Jenny M, Collombat P, Mansouri A, Tomasetto C, Madsen OD, Mellitzer G, Gradwohl G, Serup P. Genetic determinants of pancreatic epsilon-cell development. Dev Biol 2005. 286(1):217-224. [DOD] [CrossRef]
- Pedersen JK, Nelson SB, Jorgensen MC, Henseleit KD, Fujitani Y, Wright CV, Sander M, Serup P. Endodermal expression of Nkx6 genes depends differentially on Pdx1. Dev Biol 2005. 288:487-501. [DOD] [CrossRef]
- Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev 2003. 17:2591-2603. [DOD] [CrossRef]
- Matsuoka T, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol 2003. 23:6049-6062. [DOD] [CrossRef]
- Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci USA 2002. 99:6737-6742. [DOD] [CrossRef]
- Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. J Biol Chem 2002. 277:49903-49910. [DOD] [CrossRef]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, Kudo T, Engel JD, Yamamoto M, Takahashi S. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol 2005. 25:4969-4976. [DOD] [CrossRef]
- Kaneto H, Matsuoka T, Nakatani Y, Miyatsuka T, Matsuhisa M, Hori M, Yamasaki Y. A crucial role of MafA as a novel therapeutic target for diabetes. J Biol Chem 2005. 280:15047-15052. [DOD] [CrossRef]
- Matsuoka T, Kaneto H, Stein R, Miyatsuka T, Kawamori D, Henderson E, Kojima I, Matsuhisa M, Hori M, Yamasaki Y. MafA regulates expression of genes important to islet beta cell function. Mol Endocrinol 2007. 21:2764-2774. [DOD] [CrossRef]
- Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 2001. 292:1389-1394. [DOD] [CrossRef]
- Soria B, Roche E, Berna G, Leon-Quinto T, Reig JA, Martin F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 2000. 49:157-162. [DOD] [CrossRef]
- Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki K, Tzukerman M. Insulin production by human embryonic stem cells. Diabetes 2001. 50:1691-1697. [DOD] [CrossRef]
- Moritoh Y, Yamato E, Yasui Y, Miyazaki S, Miyazaki J. Analysis of insulin-producing cells during in vitro differentiation from feeder-free embryonic stem cells. Diabetes 2003. 52:1163-1168. [DOD] [CrossRef]
- Hori Y, Rulifson IC, Tsai BC, Heit JJ, Cahoy JD, Kim SK. Growth inhibitors promote differentiation of insulin-producing tissue from embryonic stem cells. Proc Natl Acad Sci USA 2002. 99:16105-16110. [DOD] [CrossRef]
- Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets expanded ductal tissue. Proc Natl Acad Sci USA 2000. 97:7999-8004. [DOD] [CrossRef]
- Taniguchi H, Yamato E, Tashiro F, Ikegami H, Ogihara T, Miyazaki J. Beta-cell neogenesis induced by adenovirus-mediated gene delivery of transcription factor pdx-1 into mouse pancreas. Gene Ther 2003. 10:15-23. [DOD] [CrossRef]
- Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB, Cornelius JG. Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Med 2000. 6:278-282. [DOD] [CrossRef]
- Zulewsky H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Muller B, Vallejo M, Thomas MK, Habener JF. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001. 50:521-533. [DOD] [CrossRef]
- Seaberg RM, Smukler SR, Kieffer TJ, Enikolopov G, Asghar Z, Wheeler MB, Korbutt G, van der Kooy D. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat Biotechnol 2004. 22:1115-1124. [DOD] [CrossRef]
- Yang L, Li S, Hatch H, Ahrens K, Cornelius JG, Petersen BE, Peck AB. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci USA 2002. 99:8078-8083. [DOD] [CrossRef]
- Yamada S, Yamamoto Y, Nagasawa M, Hara A, Kodera T, Kojima I. In vitro transdifferentiation of mature hepatocytes into insulin-producing cells. Endocr J 2006. 53:789-795. [DOD] [CrossRef]
- Ianus A, Holz GG, Theise ND, Hussain MA. In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J Clin Invest 2003. 111:843-850. [DOD]
- Suzuki A, Nakauchi H, Taniguchi H. Prospective isolation of multipotent pancreatic progenitors using flow-cytometric cell sorting. Diabetes 2004. 53:2143-2152. [DOD] [CrossRef]
- Horb ME, Shen CN, Tosh D, Slack JM. Experimental conversion of liver to pancreas. Curr Biol 2003. 13(2):105-115. [DOD] [CrossRef]
- Noguchi H, Matsushita M, Matsumoto S, Lu YF, Matsui H, Bonner-Weir S. Mechanism of PDX-1 protein transduction. Biochem Biophys Res Commun 2005. 332:68-74. [DOD] [CrossRef]
- Wu H, Macfarlane WM, Tadayyon M, Arch JR, James RF, Docherty K. Insulin stimulates pancreatic-duodenal homoeobox factor-1 (PDX1) DNA-binding activity and insulin promoter activity in pancreatic beta cells. Biochem J 1999. 344:813-818. [DOD] [CrossRef]
- Macfarlane WM, Shepherd RM, Cosgrove KE, James RF, Dunne MJ, Docherty K. Glucose modulation of insulin mRNA levels is dependent on transcription factor PDX-1 and occurs independently of changes in intracellular Ca2+. Diabetes 2000. 49(3):418-423. [DOD] [CrossRef]
- Elrick LJ, Docherty K. Phosphorylation-dependent nucleocytoplasmic shuttling of pancreatic duodenal homeobox-1. Diabetes 2001. 50:2244-2252. [DOD] [CrossRef]
- McKinnon CM, Docherty K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001. 44:1203-1214. [DOD] [CrossRef]
- Reimer MK, Ahren B. Altered beta-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57BL/6J mice. Diabetes 2002. 51:S138-S143. [DOD] [CrossRef]
- Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 2001. 414:788-791. [DOD] [CrossRef]
- Gannon M, Gamer LW, Wright CV. Regulatory regions driving developmental and tissue-specific expression of the essential pancreatic gene pdx1. Dev Biol 2001. 238(1):185-201. [DOD] [CrossRef]
- Gerrish K, Gannon M, Shih D, Henderson E, Stoffel M, Wright CV, Stein R. Pancreatic beta cell-specific transcription of the pdx-1 gene. The role of conserved upstream control regions and their hepatic nuclear factor 3beta sites. J Biol Chem 2000. 275:3485-3492. [DOD] [CrossRef]
- Shih DQ, Heimesaat M, Kuwajima S, Stein R, Wright CV, Stoffel M. Profound defects in pancreatic beta-cell function in mice with combined heterozygous mutations in Pdx-1, Hnf-1alpha, and Hnf-3beta. Proc Natl Acad Sci USA 2002. 99:3818-3823. [DOD] [CrossRef]
- Marshak S, Benshushan E, Shoshkes M, Havin L, Cerasi E, Melloul D. Functional conservation of regulatory elements in the pdx-1 gene: PDX-1 and hepatocyte nuclear factor 3beta transcription factors mediate beta-cell-specific expression. Mol Cell Biol 2000. 20(20):7583-7590. [DOD] [CrossRef]
- Gerrish K, Cissell MA, Stein R. The role of hepatic nuclear factor 1alpha and PDX-1 in transcriptional regulation of the pdx-1 gene. J Biol Chem 2001. 276:47775-47784. [DOD]
- Ben-Shushan E, Marshak S, Shoshkes M, Cerasi E, Melloul D. A pancreatic beta-cell-specific enhancer in the human PDX-1 gene is regulated by hepatocyte nuclear factor 3beta (HNF-3beta), HNF-1alpha, and SPs transcription factors. J Biol Chem 2001. 276:17533-17540. [DOD] [CrossRef]
- Samaras SE, Cissell MA, Gerrish K, Wright CV, Gannon M, Stein R. Conserved sequences in a tissue-specific regulatory region of the pdx-1 gene mediate transcription in pancreatic beta cells: Role for hepatocyte nuclear factor 3beta and Pax6. Mol Cell Biol 2002. 22:4702-4713. [DOD] [CrossRef]
- Samaras SE, Zhao L, Means A, Henderson E, Matsuoka TA, Stein R. The islet beta cell-enriched RIPE3b1/Maf transcription factor regulates pdx-1 expression. J Biol Chem 2003. 278:12263-12270. [DOD] [CrossRef]
- Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut transcription factor HNF-6 (OC-1) is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Dev Biol 2003. 258:105-116. [DOD] [CrossRef]
- Wu KL, Gannon M, Peshavaria M, Offield MF, Henderson E, Ray M, Marks A, Gamer LW, Wright CV, Stein R. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol Cell Biol 1997. 17:6002-6013. [DOD]
- Miyatsuka T, Matsuoka T, Shiraiwa T, Yamamoto T, Kojima I, Kaneto H. Ptf1a and RBP-J cooperate in activating Pdx1 gene expression through binding to Area III. Biochem Biophys Res Commun 2007. 362:905-909. [DOD] [CrossRef]
- Obata J, Yano M, Mimura H, Goto T, Nakayama R, Mibu Y, Oka C, Kawaichi M. p48 subunit of mouse PTF1 binds to RBP-Jkappa/CBF-1, the intracellular mediator of Notch signalling, and is expressed in the neural tube of early stage embryos. Genes Cells 2001. 6(4):345-360. [DOD] [CrossRef]
- Beres TM, Masui T, Swift GH, Shi L, Henke RM, MacDonald RJ. PTF1 is an organ-specific and Notch-independent basic helix-loop-helix complex containing the mammalian Suppressor of Hairless (RBP-J) or its paralogue, RBP-L. Mol Cell Biol 2006. 26:117-130. [DOD] [CrossRef]
- Krapp A, Knofler M, Ledermann B, Burki K, Berney C, Zoerkler N, Hagenbuchle O, Wellauer PK. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev 1998. 12:3752-3763. [DOD] [CrossRef]
- Fujikura J, Hosoda K, Iwakura H, Tomita T, Noguchi M, Masuzaki H, Tanigaki K, Yabe D, Honjo T, Nakao K. Notch/Rbp-j signaling prevents premature endocrine and ductal cell differentiation in the pancreas. Cell Metab 2006. 3:59-65. [DOD] [CrossRef]
- Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, Gloyn AL, Edghill EL, Hattersley AT, Wellauer PK, Goodwin G, Houlston RS. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet 2004. 36:1301-1305. [DOD] [CrossRef]
- Mashima H, Yamada S, Tajima T, Seno M, Yamada H, Takeda J, Kojima I. Genes expressed during the differentiation of pancreatic AR42J cells into insulin-secreting cells. Diabetes 1999. 48:304-309. [DOD] [CrossRef]
- Kitamura R, Ogata T, Tanaka Y, Motoyoshi K, Seno M, Takei I, Umezawa K, Kojima I. Conophylline and betacellulin-delta4: an effective combination of differentiation factors for pancreatic beta cells. Endocr J 2007. 54:255-264. [DOD] [CrossRef]
- Wiebe PO, Kormish JD, Roper VT, Fujitani Y, Alston NI, Zaret KS, Wright CV, Stein RW, Gannon M. Ptf1a binds to and activates area III, a highly conserved region of the Pdx1 promoter that mediates early pancreas-wide Pdx1 expression. Mol Cell Biol 2007. 27(11):4093-4104. [DOD] [CrossRef]
- Fujitani Y, Fujitani S, Boyer DF, Gannon M, Kawaguchi Y, Ray M, Shiota M, Stein RW, Magnuson MA, Wright CV. Targeted deletion of a cis-regulatory region reveals differential gene dosage requirements for Pdx1 in foregut organ differentiation and pancreas formation. Genes Dev 2006. 20:253-266. [DOD] [CrossRef]
- Song SY, Gannon M, Washington MK, Scoggins CR, Meszoely IM, Goldenring JR, Marino CR, Sandgren EP, Coffey RJ Jr, Wright CV, Leach SD. Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor alpha. Gastroenterology 1999. 117:1416-1426. [DOD] [CrossRef]
- Koizumi M, Doi R, Toyoda E, Masui T, Tulachan SS, Kawaguchi Y, Fujimoto K, Gittes GK, Imamura M. Increased PDX-1 expression is associated with outcome in patients with pancreatic cancer. Surgery 2003. 134:260-266. [DOD] [CrossRef]
- Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R, Jensen J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 2005. 128:728-741. [DOD] [CrossRef]
- Miyatsuka T, Kaneto H, Shiraiwa T, Matsuoka T, Yamamoto K, Kato K, Nakamura Y, Akira S, Takeda K, Kajimoto Y, Yamasaki Y, Sandgren EP, Kawaguchi Y, Wright CV, Fujitani Y. Persistent expression of PDX-1 causes acinar-to-ductal transition through Stat3 activation. Genes Dev 2006. 20(11):1435-1440. [DOD] [CrossRef]
- Soria B, Roche E, Berna G, Leon-Quinto T, Reig JA, Martin F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 2000. 49:157-162. [DOD] [CrossRef]
- Sharma A, Olson LK, Robertson RP, Stein R. The reduction of insulin gene transcription in HIT-T15 beta cells chronically exposed to high glucose concentration is associated with the loss of RIPE3b1 and STF-1 transcription factor expression. Mol Endocrinol 1995. 9:1127-1134. [DOD] [CrossRef]
- Olson LK, Redmon JB, Towle HC, Robertson RP. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J Clin Invest 1993. 92:514-519. [DOD] [CrossRef]
- Poitout V, Olson LK, Robertson RP. Chronic exposure of betaTC-6 cells to supraphysiologic concentrations of glucose decreases binding of the RIPE3b1 insulin gene transcription activator. J Clin Invest 1996. 97:1041-1046. [DOD] [CrossRef]
- Jonas JC, Sharma A, Hasenkamp W, Iikova H, Patane G, Laybutt R, Bonner-Weir S, Weir GC. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem 1999. 274:14112-14121. [DOD] [CrossRef]
- Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest 1992. 90:320-325. [DOD] [CrossRef]
- Dandona P, Thusu K, Cook S, Snyder B, Makowski J, Armstrong D, Nicotera T. Oxidative damage to DNA in diabetes mellitus. Lancet 1996. 347:444-445. [DOD]
- Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 1999. 48:1-9. [DOD] [CrossRef]
- Ihara Y, Toyokuni S, Uchida K, Odaka H, Tanaka T, Ikeda H, Hiai H, Seino Y, Yamada Y. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 1999. 48:927-932. [DOD] [CrossRef]
- Matsuoka T, Kajimoto Y, Watada H, Kaneto H, Kishimoto M, Umayahara Y, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest 1997. 99:144-150. [DOD] [CrossRef]
- Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y, Hori M. Beneficial effects of antioxidants for diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999. 48:2398-2406. [DOD] [CrossRef]
- Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci USA 1999. 96(19):10857-10862. [DOD] [CrossRef]
- Kajimoto Y, Matsuoka T, Kaneto H, Watada H, Fujitani Y, Kishimoto M, Sakamoto K, Matsuhisa M, Kawamori R, Yamasaki Y, Hori M. Induction of glycation suppresses glucokinase gene expression in HIT-T15 cells. Diabetologia 1999. 42:417-1424. [DOD] [CrossRef]
- Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function by provoking oxidative stress. J Biol Chem 2001. 276:31099-31104. [DOD] [CrossRef]
- Tanaka Y, Tran PO, Harmon J, Robertson RP. A role of glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci USA 2002. 99:12363-12368. [DOD] [CrossRef]
- Gorogawa S, Kajimoto Y, Umayahara Y, Kaneto H, Watada H, Kuroda A, Kawamori D, Yasuda T, Matsuhisa M, Yamasaki Y, Hori M. Probucol preserves pancreatic beta-cell function through reduction of oxidative stress in type 2 diabetes. Diabetes Res Clin Prac 2002. 57:1-10. [DOD] [CrossRef]
- Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003. 52:581-587. [DOD] [CrossRef]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003. 52:1-8. [DOD]
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003. 144(12):5159-5165. [DOD] [CrossRef]
- Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes - a convergence of glucotoxicity and lipotoxicity. Endocrinology 2002. 143(2):339-342. [DOD] [CrossRef]
- Moore PC, Ugas MA, Hagman DK, Parazzoli SD, Poitout V. Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes 2004. 53:2610-2616. [DOD] [CrossRef]
- Hagman DK, Hays LB, Parazzoli SD, Poitout V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J Biol Chem 2005. 280:32413-32418. [DOD] [CrossRef]
- Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamura K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M, Araki E. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun 2003. 300:216-222. [DOD] [CrossRef]
- Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem 2004. 271:42351-42354. [DOD] [CrossRef]
- Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka T. Involvement of oxidative stress and the JNK pathway in glucose toxicity. Rev Diabet Stud 2004. 1(4):165-174. [DOD] [CrossRef]
- Harmon JS, Stein R, Robertson RP. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 2005. 280:11107-11113. [DOD] [CrossRef]
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Rad Biol Med 1996. 20:463-466. [DOD] [CrossRef]
- Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997. 46:1733-1742. [DOD] [CrossRef]
- Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 2002. 277:30010-30018. [DOD] [CrossRef]
- Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, Watada H, Leibiger IB, Yamasaki Y, Hori M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun N-terminal kinase. Diabetes 2003. 52:2896-2904. [DOD] [CrossRef]
- Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 2002. 110:1839-1847. [DOD]
- Nakae J, Biggs WH 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 2002. 32(2):245-253. [DOD] [CrossRef]
- Kawamori D, Kaneto H, Nakatani Y, Matsuoka T, Matsuhisa M, Hori M, Yamasaki Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J Biol Chem 2006. 281:1091-1098. [DOD] [CrossRef]
This article has been cited by other articles:

|
Pancreatic stem/progenitor cells for the treatment of diabetes
Noguchi H
Rev Diabet Stud 2010. 7(2):105-111
|
|

|
The Expression Pattern of PDX-1, SHH, Patched and Gli-1 Is Associated with Pathological and Clinical Features in Human Pancreatic Cancer
Quint K, Stintzing S, Alinger B, Hauser-Kronberger C, Dietze O, Gahr S, Hahn EG, Ocker M, Neureiter D
Pancreatology 2008. 9(1-2):116-126
|
|
|


)
)
)
)
)
)
)

