Review
| Rev Diabet Stud,
2008,
5(3):136-143 |
DOI 10.1900/RDS.2008.5.136 |
The Current Status of Islet Transplantation and its Perspectives
Naoya Kobayashi
Department of Surgery, Okayama University Graduate School of Medicine and Dentistry, 2-5-1 Shikata-cho, Okayama 700-8558, Japan, e-mail: immortal@md.okayama-u.ac.jp
Manuscript submitted July 20, 2008; resubmitted November 13, 2008; accepted November 18, 2008.
Keywords: diabetes, islet transplantation, beta-cell regeneration, stem cell
Abstract
Transplantation of human pancreatic isolated islets can restore beta-cell function but it requires chronic immunosuppression. The outcome of islet transplantation mainly depends on both the quality of islet preparations, and the survival of the graft. The quality of islet preparations can be evaluated by the results of isolation, which determines the chance to achieve insulin independence. The survival of islet grafts is reflected by the amount of engrafted functional tissue that maintains metabolic control. Immunosuppressive therapy prevents the immunological rejection of grafts, but impairs their function and impedes their regenerative capacity. Therefore, the selection of high quality islet preparations and the reduction of toxic effects of immunosuppressive regimens might dramatically improve the outcomes. The application of stem cell therapy in islet transplantation may contribute to a better understanding of the mechanisms responsible for tissue homeostasis and immune tolerance. Xenogeneic islets may serve as an unlimited source if immune tolerance can be achieved. This may be a strategy to enable a substantial improvement in function while overcoming potentially deleterious risks.
Introduction
Diabetes mellitus represents a major cause of death and disability worldwide. It is a chronic disease characterized by a failure of glucose homeostasis, which may lead to kidney failure, retinopathy, and neuropathy, and shortens life expectancy. Type 1 diabetes results from autoimmune destruction of insulin-producing cells. Therefore, patients require exogenous administration of insulin to survive. In contrast, type 2 diabetes results from a progressive impaired insulin secretion associated with peripheral insulin resistance. The chronic development of such events may exhaust the pancreatic regenerative reserve, thus resulting in the need to administer exogenous insulin in the advanced stages [1].
Historical perspective
Insulin therapy for diabetes has been utilized over the last 75 years, since the first patient was treated in 1922. However, it does not constitute a cure and cannot prevent the onset of chronic deleterious complications [2]. In 1967, Lacy introduced the idea of islet transplantation [3]. However, it did not become a successful reality for human treatment until 2000, when Shapiro and associates introduced the Edmonton protocol, collecting islets from 2-4 donor pancreata in combination with a glucocorticoid-free immunosuppressive regimen [4]. The results after the Edmonton protocol established a landmark towards a cure for diabetes. Other remarkable breakthroughs during the 1990’s were the discovery of glucagon-like peptide-1 (GLP-1) by Holz and associates in 1993 [5], and the elucidation of the new role that C-peptide plays in preventing the development of vascular and neural dysfunction in diabetes by Ido and co-workers in 1997 [6].
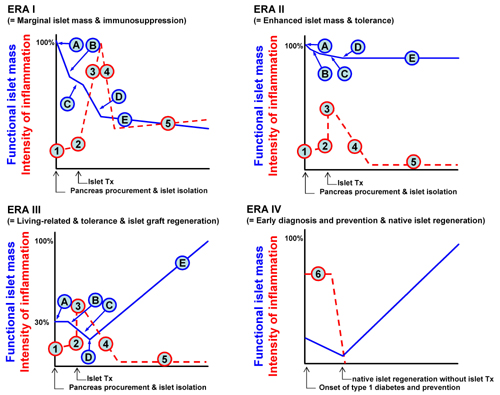
Later, the main focus of islet transplantation changed towards the concept of “eras”, as introduced by Ricordi and colleagues [7] (Figure 1). The Edmonton protocol would therefore be more appropriate to fit in Era I, as described in the figure sequence. Points A and B represent the islet loss after isolation, and thereby, the use of multiple donors to compensate for it. Point C, represents the transient islet culture that is utilized to remove undesired tissue (i.e. dead cells, exocrine cells and donor leukocytes), and to initiate immunosuppression. Point D in Figure 1 stands for post-transplant early islet loss, which happens due to the instant blood-mediated inflammatory reaction (IBMIR) and a phenomenon described as “portal storm”. The latter means an elevated immunosuppressant level in portal blood compared with the systemic level after administration in the portal vein [8]. Finally, in point E the islet grafts must stand between the chronic rejection/recurrence of autoimmune disease and the toxic side effects of the immunosuppressive agents.
 |
 |
Figure 1. The concept of eras in islet transplantation. In ERA I, points A and B represent apoptotic islets lost after isolation. C represents the transient islet culture to remove most of the undesired tissue, (dead cells, exocrine cells and donor leukocytes) but also to initiate immunosuppression. D represents the early post-transplant islet loss due to instant blood-mediated inflammatory reaction (IMBIR), and "portal storm". Finally, in E, islet grafts must stand between the chronic rejection/recurrence of autoimmune disease and the toxic side effects of the immunosuppressive regimen. In ERA II, maintenance of islet mass could be achieved by an efficient isolation method and substitution of immunosuppressants. In ERA III, a perfect islet isolation method should be developed and regeneration of islet grafts should be facilitated by using regenerative medicine. Therefore, one third of the islet mass in ERA I allows us to treat one diabetic patient. In this setting, a living-related islet transplantation will spread out world wide. Otherwise, one donor pancreas can be used to treat multiple patients. Ultimately, early diagnosis of type 1 diabetes onset and induction of beta-cell tolerance could allow pancreatic regeneration without pancreas or islet transplantation. "Time" for the X axis, "functional islet mass" and "intensity of inflammation" on the Y axis. Tx: transplantation. A: pancreas procurement and preservation. B: yield of isolated islets. C: early loss of the islets by innate immunity. D: engrafted islets. E: survived islets. 1: basal level of inflammation. 2: trigger of innate immunity. 3: adapted immunity. 4: graft accommodation. 5: chronic rejection (ERA I) or tolerance (EAR II and III). 6: occurrence of autoimmunity. Figure modified according to Ricordi et al. [7]. |
|
In 2005, a new breakthrough by Matsumoto and associates, was generated by the introduction of an improved islet isolation method, now referred to as the Kyoto Method [9]. In Japan, the availability of pancreata for clinical cadaveric islet transplantation is restricted to non-heart-beating donors (NHBDs). Therefore, Matsumoto and their colleagues needed to modify the current standard islet isolation protocol for brain-dead donors, and make it suitable for NHBDs. The Kyoto islet isolation method is the one with several steps of induction based on the ideas already reported, and originally invented by their group. Using the Kyoto method, they isolated islets from 13 human pancreata of NHBDs, and transplanted 11 preparations to six type-1 diabetic patients. The Kyoto method was also the world’s first living-donor islet transplantation. The rate to meet the Edmonton protocol release criteria was 84.6%. The improved isolation method and the use of a living donor resulted in a larger number of harvested islets per gram of donor pancreatic tissue [10]. The manipulation of those variables revealed in part the aspects that would lead to Era II-III of islet transplantation (Figure 1), whilst the work on tolerance remained to be completed. Therefore, in principle, the goal for islet cell replacement (Era II-III) is a supply of functional beta-cells with the capacity of glucose-responsiveness (mature phenotype) to secrete insulin into the blood stream in a physiological manner. Secondly, a functional engraftment must be achieved to promote beta-cell regeneration while it balances with beta-cell turnover. In other words, a positive balance is needed between factors that promote beta-cell replication or stem cell differentiation within islet grafts, and a minimally toxic immunosuppressive regimen which allows beta-cell proliferation.
In reality, the current practices for islet transplantation in humans do not work satisfactorily. One possible reason for only partial success is the choice of transplantation site. There is evidence that the liver is a less than optimal environment for islets and that it contributes to short- and long-term beta-cell destruction or failure [11]. A current study has demonstrated that the pancreas is a more suitable transplant site, and requires fewer islets than standard sites, such as the liver or kidneys. Also, it leads to improvements in transplantation outcomes in both rats and dogs [11]. Thus, the authors have concluded that the pancreas should be considered as an islet transplant site in humans.
The normal pancreatic beta-cell homeostasis
There are several observations regarding mechanisms that support stem cell differentiation, beta-cell replication and the homeostasis of pancreatic beta-cell mass [12-15]. The endocrine pancreas represents a minute fraction, about 1% of the pancreatic mass, but is highly vascularized, since each islet may receive a blood supply from 2-3 arterioles and drainage into 1-2 venules [12]. beta-cells seem to have a close relationship with the vessels since their metabolic activity demands high energy utilization and requires synchronization to release large amounts of insulin in a short time [12, 13]. Due to their high level of activity, beta-cells turnover has been estimated at 40-50 days in rodents [14]. Therefore, beta-cells continuously undergo apoptosis at the end of their life span, albeit quite slowly, when they are possibly replaced by newly generated "mesenchymal-epithelial transition" cells from the ducts. After stimulation, intra-islet progenitors can generate newly formed beta-cells within 2-3 days [15].
There are reports indicating that either ductal cells, or intra-islet cell progenitors, can produce insulin-producing cells, as well as other pancreatic cell types, in vitro [15-17]. In experiments, both cell types could be expanded to some extent during in vitro culture, and then redifferentiated into insulin-producing cells. However, the amounts of insulin released, and their glucose-responsiveness, seem to be reduced when compared with normal isolated islets [15, 16]. Also, evidence has been found of a transition from ductal to insulin-producing cells, which formed solitary clumps as a result of peripheral insulin resistance, pregnancy, pancreatic duct ligation or pancreatectomy [3].
On the other hand, two elegant studies have shown evidence of beta-cell replication for the maintenance of beta-cell homeostasis. The first one was performed by the use of a cell tracking method utilizing an alkaline phosphatase protein labeling system, controlled by an active transcription of the insulin gene. In this study, the generation of beta-cell progeny was identified by the expression of the labeling marker once the cells were derived from mature beta-cells, but not from other cell types [18]. The second study examined the role of pancreatic regeneration after the induction of apoptosis in 70-80% of the beta-cells. This group used conditionally ablated Ins-rt-TET/DTA (Tetracycline/Diphteria toxin A) double transgenic mice, in which the administration of doxycycline triggered diabetes. The regenerative capacity of islets was tested by withdrawing doxycycline, and resulted in euglycemia with almost complete recovery of the normal islet architecture. Interestingly, when tacrolimus and sirolimus (two immunosuppressive drugs use in the Edmonton protocol) were subsequently administrated after doxycycline withdrawal, they completely prevented beta-cell regeneration in diabetic mice [2, 19].
The abovementioned studies strongly suggest that endogenous beta-cell regeneration may be achieved if the autoimmune disease can be halted early, or soon after diabetes onset, by "regeneration-compatible drugs" [2, 19]. This strategy is an essential component of Era IV in islet transplantation. However, patients who have exhausted their regenerative capacity by collapse of residual stem cells, or replicating beta-cells, would be better candidates for islet transplantation in combination with the toleragenic immunosuppressive drugs (as referred to the drugs in Era III, Figure 1), which could act to be "regeneration-compatible".
Factors promoting beta-cell regeneration in situ and after islet transplantation
As previously described, the endocrine pancreas is subjected to dynamic changes in response to variations in the global metabolic demands. Therefore, it is important to interpret those mechanisms mediated through hormones and growth factors, which serve as effectors to keep beta-cell formation (neogenesis and proliferation) and beta-cell death (senescence, apoptosis and necrosis) in balance. Amongst the main factors promoting beta-cell proliferation are nutrients, hormones and growth factors [1, 11].
In this regard, it is interesting that glucose increases the number of beta-cells by approximately 50% when it is infused in rats during a 24 h period [13, 20]. In contrast, when isolated islets are maintained in the presence of high glucose, they may even release interleukin-1β (IL-1β) and undergo apoptosis [20, 21]. Insulin itself acts as a potent inducer of hyperplasia and hypertrophy via insulin receptor substrate-1 (IRS-1) genes, whilst providing positive feedback to beta-cells as a compensatory change at the early stage of peripheral insulin resistance. Genetically engineered mice, which lacked insulin receptors specifically in beta-cells, exhibited a considerably decreased beta-cell mass and developed diabetes [15, 22].
Researchers have identified insulin-like growth factor-II (IGF-II) as an essential element to maintain the growth of fetal and neonatal islets. It is mainly secreted by the surrounding ductal cells [23]. The use of IGF-II in islet transplantation allowed the suppression of islet apoptosis and promoted islet proliferation [24]. Likewise, fibroblast growth factor-2 (FGF-2) is a multi-regulatory factor, which can induce proliferation of both progenitor and beta-cells [1, 15, 25-27]. When FGF-2 knockout mice were generated, an in vivo attenuated FGF-2 signaling led to diabetes in mice [26]. We have previously reported the use FGF-2 as an inductor of regeneration, and we have found facilitated revascularization of islet grafts after transplantating a marginal islet mass [27].
Hepatocyte growth factor (HGF), mainly secreted by pancreatic endothelial cells, is also able to induce beta-cell proliferation in islets and to cause insulinotropic effects in fetal islets. An excellent study with transgenic mice overexpressing HGF under the control of rat insulin promoter (RIP), demonstrated a dramatic increase in islet mass, while reflecting an enlarged size of islets and an increased beta-cell number [28]. Moreover, when HGF was intraperitoneally administrated, the transplantation of a marginal dose of pancreatic islets resulted in the amelioration of diabetes in mice [29].
GLP-1 is a potent intestinal insulinotropic hormone and seems to be useful to lower glucose levels in rodents, and in human diabetic subjects of types 1 and 2. However, GLP-1 has a very short half-life of 2-6 min, which hampers its therapeutic application. Therefore, researchers are now taking advantage of the availability of recombinant analog exendin-4, which is more potent than GLP-1, and possesses a much longer half-life of 2-6 h for in vivo studies. Studies on exendin-4 have clearly demonstrated multiple benefits, in type 2 diabetic patients, and in STZ-induced diabetic mice, whilst receiving transplantation of a marginal dose of islets. Observed benefits include enhancement of the beta-cell functionality, replication and neogenesis. [30, 31]. The mechanisms of islet renewal and the effects of these growth factors have been studied mainly in rodents. Therefore, it may not be directly relevant to humans.
Control of quality in transplantable islet cells
Normal isolated islets are indeed the cell prototype for functional comparison. Considering islet equivalent (IEQ) as a unit of transplantable function, when a larger number of islets are transplanted, the serum levels of C-peptide increase and the exogenous insulin requirement decreases, resulting in a greater probability of insulin independence [32]. However, large variations have been observed in the potency and number of beta-cells among islet preparations. These factors at work, include beta-cell content/count, viability, integrity and inflammatory processes, and they compromise the probability of successful islet engraftment. Therefore, a multi-factorial quality assessment of islet preparations including a reliable count of viable islets is required to assure consistent benefits after islet transplantation [32, 33].
Potency of transplantable islet cells
Potency tests of islet preparations conventionally involve two major aspects. The first is glucose-responding insulin secretion. This is often expressed as an index of the ratio between basal low glucose incubation and stimulation delivered with glucose loading, secretagogues, or drugs under static incubation or dynamic perfusion.
It is worth noting that islets, which preserve a well-defined morphology and are constituted with a high amount of viable cells, may present a low level of glucose-responsive insulin secretion, but that those islets may be capable of restoring normoglycemia after transplantation [32]. These findings strongly suggests that an insulin secretory defect caused by ongoing apoptosis, inflammatory processes, osmolar damage, cell exhaustation and transitory energy depletion is reversible if the noxious stimuli are well controlled in the islets [34]. Therefore, interventions, which enable a functional recovery of islets, may enhance their potential, and preferably expand the transplantable islet mass [32, 34].
The second aspect in testing islet potency is the use of tests for diabetes reversal in immunodeficient nude mice. In this model, transplantation of isolated islets is correlated with the capacity of engraftment and the potential to restore islet function in an islet dosage- and time-dependent manner. Despite the accuracy of bioassay, it does not enable a practical selection of the preparations for islet transplantation because of its time-consuming aspect [32]. Ichii and associates have developed a method for more accurate assessment of the islet composition and beta-cell content in islet preparations at two different time points [35]. Their method discriminates dead cells from living cells using 7AAD staining and simultaneous TMRE staining for mitochondrial membrane potential in both beta- and non-beta-cells. The measured number of viable beta-cells within an islet preparation correlates well with the results of bioassay to predict the reversal of diabetes, and is defined as islet potency.
Safety of transplantable islet cells
The safety of transplantable islet cells has to be determined by their quality in terms of potency, as mentioned above, and the extent to which potency may contribute to transplantable functionality. However, other types of cells contained in islet preparations are also typically transplanted to the recipient [32, 36]. Therefore, it is preferable to remove cells that may trigger early or immediate immunological responses to beta-cells as a target of the host immune system. One of the triggers can be the presence of passenger leukocytes, which express several proinflammatory molecules such as monocyte chemoattractant protein-1 (MCP-1), tissue factor, IL-1beta and others. Donor passenger leukocytes can be removed by culturing islet preparations in vitro prior to transplantation [36].
Alternatives to isolated islets
The worldwide shortage of human donor pancreata for islet cell transplantation has led to search for alternative sources of islet cells including stem cell-derived insulin secreting cells, replicated beta-cells, artificial cells and xenogeneic islets. However, such sources possess natural advantages and disadvantages that need to be strictly assessed, before they can serve as a therapeutic source.
Stem cell-derived insulin secreting cells
In principle, stem cells have self-renewal and variable differentiating capacities when they reside in their niches, thereby participating in tissue homeostasis. However, phenotype and localization of such stem cells within the postnatal pancreas remain to be fully elucidated, as mentioned above [13]. In an attempt to identify an alternative source of mature isolated islets, researchers have conducted numerous studies on isolation and differentiation of putative pancreatic stem cells (ductal and intra-islet progenitors) and other stem cells, including embryonic stem (ES), bone marrow, umbilical cord and hematopoietic stem cells [3, 13-17, 37-41].
Despite the unlimited proliferation and differentiating potential of ES cells, the achievement of a functional mature phenotype was more difficult than initially expected. Insulin biosynthesis and glucose-responsiveness by cells derived from ES cells are much lower or absent compared to the single-cell function in normal pancreatic beta-cells from isolated islets [39]. The presence of undifferentiated ES cells residing after in vitro induction, increases the possibilities for tumor formation. Therefore, a better understanding of the mechanisms that control pluripotency and those which regulate the islet beta-cell differentiation is needed [42].
Experiments with bone marrow or hematopoietic stem cells fail to demonstrate a significant amount of functional transdifferentiated beta-cells. These transplanted stem cells promoted pancreatic regeneration and immune tolerance. Pancreatic regeneration was stimulated by endothelial cells derived from the engrafted stem cells, leading to an increase in the host beta-cell mass. Immunological tolerance was achieved via chimerism [43, 44]. The use of stem cells differentiated in vitro, such as insulin-secreting units, must be comparable to that of isolated islets in order to provide a sufficient transplantable function. If those newly differentiated stem cells possess a restricted low secretory function (defined as islet potency), then a large amount of cells would be required to achieve insulin independency. However, this can increase the probability of tumor formation, decrease the engraftment potential and exacerbate inflammatory signals that trigger allo-autoimmune reactions. On the other hand, defined populations of toleragenic stem cells may not contribute with transplantable function, but may indirectly promote pancreatic regeneration [1, 36].
Xenogeneic islets
The alternative use of xenogeneic tissues constitutes an attractive and virtually unlimited source for islet replacement. However, islet cell function is still limited due to physiological differences, rather than an incomplete phenotype expression. Hyperacute rejection due to natural anti-alpha Gal antibodies is one of the major hurdles in pig-to-human xenotransplantation of islets. However, porcine endocrine islet cells seem to express relatively low alpha Gal antigen. In an experiment, porcine transplanted islets were relatively tolerant of the innate immunological stress from the alpha Gal antibodies-mediated acute humoral xenograft rejection, in pig-to-macaque transplantation [45]. Nevertheless, robust immunosuppressive regimen is required to attain graft survival. In many cases, toxic side effects of immunosuppression dramatically affect graft function and increase the probability of zoonosis transmission. There is a particular concern about the potential for infections occurring in recipients due to porcine endogenous retrovirus (PERV). This imposes life-threatening risks for recipients and, contains potential for public biohazards [46].
Pancreas vs. islet transplantation in diabetic patients
Transplantation options for diabetic patients are pancreas and islet transplantation. Pancreas transplantation has been actively performed for the past 30 years with improving success rates. On the other hand, islet transplantation provides the advantage of being less invasive with fewer complications. However, current experience shows that multiple transplants are required to achieve and maintain insulin-independence transiently. Long-term function still remains a problem even with multiple transplantation. Early successes with single-donor islet transplants are encouraging and if maintained will largely substitute pancreas transplants. Currently, single-donor islet transplants have been shown to work in recipients with low insulin requirements who receive a pancreas from a donor with high body mass index. However, pancreas transplants from obese donors are associated with increased surgical risk. Therefore, it is logical to preferentially allocate obese donor pancreata to islet recipients. In addition, pancreata obtained from older donors of 50 to 65 years could be preferentially allocated to islets since their islet yield is still good, whereas they are associated with decreased survival in whole-organ pancreas transplants. With increasing efficiency and success of islet transplants, the criteria for pancreas allocation for islets should be periodically considered [47].
Conclusions
Islet transplantation was first proposed as a potential cure for diabetic patients some 40 years ago. However, specific limitations avoid the widespread use of this treatment until now. The most critical limitations are the availability of transplantable tissue and the toxic side effects of immunosuppressive regimens. The quality of islet preparations is in direct relation to the probability of success in achieving insulin independence. On the other hand, maintenance of the restored function requires tolerance, to halt immunological attack, and to allow proliferation of the grafts.
The ideal treatment for diabetic patients is islet regeneration therapy, induction of self-tolerance for early-diagnosed patients, and additional transplantation of islets to serve as a pool for tissue homeostasis. Alternative islet sources constitute attractive models for the study of these goals of tolerance and tissue regeneration. However, in terms of function and safety, such alternative sources remain unsatisfactory at present.
References
- Halban PA. Cellular sources of new pancreatic beta cells and therapeutic implications for regenerative medicine. Nat Cell Biol 2004. 6(11):1021-1025. [DOD] [CrossRef]
- Kaestner KH. Beta cell transplantation and immunosuppression: can't live with it, can't live without it. J Clin Invest 2007. 117(9):2380-2382. [DOD] [CrossRef]
- Trucco M. Regeneration of the pancreatic beta cell. J Clin Invest 2005. 115(1):5-12. [DOD]
- Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000. 343(4):230-238. [DOD] [CrossRef]
- Holz GG, Kuhtreiber WM, Habener JF. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37). Nature 1993. 361(6410):362-365. [DOD] [CrossRef]
- Ido Y, Vindigni A, Chang K, Stramm L, Chance R, Heath WF, DiMarchi RD, Di Cera E, Williamson JR. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science 1997. 277(5325):563-566. [DOD] [CrossRef]
- Ricordi C, Inverardi L, Kenyon NS, Goss J, Bertuzzi F, Alejandro R. Requirements for success in clinical islet transplantation. Transplantation 2005. 79(10):1298-1300. [DOD] [CrossRef]
- Shapiro AM, Gallant HL, Hao EG, Lakey JR, McCready T, Rajotte RV, Yatscoff RW, Kneteman NM. The portal immunosuppressive storm: relevance to islet transplantation? Ther Drug Monit 2005. 27(1):35-37. [DOD]
- Okitsu T, Matsumoto S, Iwanaga Y, Noguchi H, Nagataa H, Yonekawa Y, Maekawa T, Tanaka K. Kyoto islet isolation method: The optimized one for non-heart-beating donors with highly efficient islet retrieval. Transplant Proc 2005. 37(8):3391-3392. [DOD]
- Matsumoto S, Okitsu T, Iwanaga Y, Noguchi H, Nagata H, Yonekawa Y, Yamada Y, Fukuda K, Tsukiyama K, Suzuki H, et al. Insulin independence after living-donor distal pancreatectomy and islet allotransplantation. Lancet 2005. 365(9471):1642-1644. [DOD]
- Stagner JI, Rilo HL, White KK. The pancreas as an islet transplantation site. Confirmation in a syngeneic rodent and canine autotransplant model. J Pancreas 2007. 8(5):628-636. [DOD]
- Konstantinova I, Lammert E. Microvascular development: learning from pancreatic islets. Bioessays 2004. 26(10):1069-1075. [DOD] [CrossRef]
- Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev 2005. 85(4):1255-1270. [DOD] [CrossRef]
- Bonner-Weir S. Beta-cell turnover: its assessment and implications. Diabetes 2001. 50(Suppl 1):S20-S24. [DOD] [CrossRef]
- Zulewski H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Müller B, Vallejo M, Thomas MK, Habener JF. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001. 50(3):521-533. [DOD] [CrossRef]
- Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A 2000. 97(14):7999-8004. [DOD] [CrossRef]
- Lechner A, Habener JF. Stem/progenitor cells derived from adult tissues: potential for the treatment of diabetes mellitus. Am J Physiol Endocrinol Metab 2003. 284(2):E259-E266. [DOD]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004. 429(6987):41-46. [DOD] [CrossRef]
- Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 2007. 117(9):2553-2561. [DOD] [CrossRef]
- Bernard C, Berthault MF, Saulnier C, Ktorza A. Neogenesis vs. apoptosis As main components of pancreatic beta cell mass changes in glucose-infused normal and mildly diabetic adult rats. Faseb J 1999. 13(10):1195-1205. [DOD]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002. 110(6):851-860. [DOD]
- Otani K, Kulkarni RN, Baldwin AC, Krutzfeldt J, Ueki K, Stoffel M, Kahn CR, Polonsky KS. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am J Physiol Endocrinol Metab 2004. 286(1):E41-E49. [DOD] [CrossRef]
- Hill DJ, Strutt B, Arany E, Zaina S, Coukell S, Graham CF. Increased and persistent circulating insulin-like growth factor II in neonatal transgenic mice suppresses developmental apoptosis in the pancreatic islets. Endocrinology 2000. 141(3):1151-1157. [DOD] [CrossRef]
- Ilieva A, Yuan S, Wang RN, Agapitos D, Hill DJ, Rosenberg L. Pancreatic islet cell survival following islet isolation: the role of cellular interactions in the pancreas. J Endocrinol 1999. 161(3):357-364. [DOD] [CrossRef]
- Elghazi L, Cras-Meneur C, Czernichow P, Scharfmann R. Role for FGFR2IIIb-mediated signals in controlling pancreatic endocrine progenitor cell proliferation. Proc Natl Acad Sci U S A 2002. 99(6):3884-3889. [DOD] [CrossRef]
- Hart AW, Baeza N, Apelqvist A, Edlund H. Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature 2000. 408(6814):864-868. [DOD] [CrossRef]
- Rivas-Carrillo JD, Navarro-Alvarez N, Soto-Gutierrez A, Okitsu T, Chen Y, Tabata Y, Misawa H, Noguchi H, Matsumoto S, Tanaka N, Kobayashi N. Amelioration of diabetes in mice after single-donor islet transplantation using the controlled release of gelatinized FGF-2. Cell Transplant 2006. 15(10):939-944. [DOD] [CrossRef]
- Garcia-Ocana A, Takane KK, Syed MA, Philbrick WM, Vasavada RC, Stewart AF. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem 2000. 275(2):1226-1232. [DOD] [CrossRef]
- Nakano M, Yasunami Y, Maki T, Kodama S, Ikehara Y, Nakamura T, Tanaka M, Ikeda S. Hepatocyte growth factor is essential for amelioration of hyperglycemia in streptozotocin-induced diabetic mice receiving a marginal mass of intrahepatic islet grafts. Transplantation 2000. 69(2):214-221. [DOD] [CrossRef]
- King A, Lock J, Xu G, Bonner-Weir S, Weir GC. Islet transplantation outcomes in mice are better with fresh islets and exendin-4 treatment. Diabetologia 2005. 48(10):2074-2079. [DOD] [CrossRef]
- Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999. 48(12):2270-2276. [DOD] [CrossRef]
- Bertuzzi F, Ricordi C. Prediction of clinical outcome in islet allotransplantation. Diabetes Care 2007. 30(2):410-417. [DOD] [CrossRef]
- Girman P, Berkova Z, Dobolilova E, Saudek F. How to use image analysis for islet counting. Rev Diabet Stud 2008. 5(1):38-46. [DOD] [CrossRef]
- Rivas-Carrillo JD, Soto-Gutierrez A, Navarro-Alvarez N, Noguchi H, Okitsu T, Chen Y, Yuasa T, Tanaka K, Narushima M, Miki A, et al. Cell-permeable pentapeptide V5 inhibits apoptosis and enhances insulin secretion, allowing experimental single-donor islet transplantation in mice. Diabetes 2007. 56(5):1259-1267. [DOD] [CrossRef]
- Ichii H, Inverardi L, Pileggi A, Molano RD, Cabrera O, Caicedo A, Messinger S, Kuroda Y, Berggren PO, Ricordi C. A novel method for the assessment of cellular composition and beta-cell viability in human islet preparations. Am J Transplant 2005. 5(7):1635-1645. [DOD] [CrossRef]
- Halme DG, Kessler DA. FDA regulation of stem-cell-based therapies. N Engl J Med 2006. 355(16):1730-1735. [DOD] [CrossRef]
- Efrat S. Ex-vivo Expansion of Adult Human Pancreatic Beta-Cells. Rev Diabet Stud 2008. 5(2):116-122. [DOD] [CrossRef]
- Koblas T, Harman SM, Saudek F. The application of umbilical cord blood cells in the treatment of diabetes mellitus. Rev Diabet Stud 2005. 2(4):228-234. [DOD] [CrossRef]
- D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 2006. 24(11):1392-1401. [DOD] [CrossRef]
- Lechner A, Yang YG, Blacken RA, Wang L, Nolan AL, Habener JF. No evidence for significant transdifferentiation of bone marrow into pancreatic beta-cells in vivo. Diabetes 2004. 53(3):616-623. [DOD] [CrossRef]
- Rajagopal J, Anderson WJ, Kume S, Martinez OI, Melton DA. Insulin staining of ES cell progeny from insulin uptake. Science 2003. 299(5605):363. [DOD]
- Fujikawa T, Oh SH, Pi L, Hatch HM, Shupe T, Petersen BE. Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell-derived insulin-producing cells. Am J Pathol 2005. 166(6):1781-1791. [DOD]
- Hussain MA, Theise ND. Stem-cell therapy for diabetes mellitus. Lancet 2004. 364(9429):203-205. [DOD] [CrossRef]
- Suri A, Calderon B, Esparza TJ, Frederick K, Bittner P, Unanue ER. Immunological reversal of autoimmune diabetes without hematopoietic replacement of beta cells. Science 2006. 311(5768):1778-1780. [DOD] [CrossRef]
- Hering BJ, Wijkstrom M, Graham ML, Hardstedt M, Aasheim TC, Jie T, Ansite JD, Nakano M, Cheng J, Li W, et al. Prolonged diabetes reversal after intraportal xenotransplantation of wild-type porcine islets in immunosuppressed nonhuman primates. Nat Med 2006. 12(3):301-303. [DOD] [CrossRef]
- Paradis K, Langford G, Long Z, Heneine W, Sandstrom P, Switzer WM, Chapman LE, Lockey C, Onions D, Otto E. Search for cross-species transmission of porcine endogenous retrovirus in patients treated with living pig tissue. The XEN 111 Study Group. Science 1999. 285(5431):1236-1241. [DOD] [CrossRef]
- Kandaswamy R, Sutherland D. Pancreas versus islet transplantation in diabetes mellitus: how to allocate deceased donor pancreata? Transplant Proc 2006. 38(2):365-367. [DOD]
This article has been cited by other articles:
|


)


