Original Data
| Rev Diabet Stud,
2009,
6(1):43-53 |
DOI 10.1900/RDS.2009.6.43 |
GAD65-Specific Cytotoxic T Lymphocytes Mediate Beta-Cell Death and Loss of Function
Sarah Rasche, Rhea Y. Busick, Anthony Quinn
Department of Biological Sciences, University of Toledo, 2801 W. Bancroft, Toledo, OH 43606, USA
Address correspondence to: Anthony Quinn, e-mail: aquinn@utnet.utoledo.edu
Manuscript submitted March 12, 2009; resubmitted May 12, 2009; accepted May 20, 2009.
Keywords: type 1 diabetes, T cell, GAD65, major histocompatibility complex class I, MHC, NOD.scid, antigen, FasL, beta-cell, pathogenesis
Abstract
Autoimmunity to islet cell antigens like glutamic acid decarboxylase 65kD (GAD65) is associated with the destruction of insulin-producing β-cells and progression to type 1 diabetes (T1D) in NOD mice and humans. T cell responses to GAD65 are detectable in the spleen of prediabetic NOD mice and in the peripheral blood of humans prior to the onset of overt hyperglycemia. Previous findings from our lab revealed that GAD65546-554-specific cytotoxic T lymphocytes (CTL) are present in naïve NOD mice and are able to induce islet inflammation upon adoptive transfer into NOD.scid recipients. Additionally, we found that professional antigen-presenting cells (APC) generate the p546-554 epitope from a soluble GAD65 fragment, p530-554, and from GAD65 released by injured β-cells in vivo. Here, we report that the GAD65 fragment p546-554 is a dominant CTL-inducing epitope which is naturally processed and presented by a GAD65-expressing β-cell line. Further, co-culture of GAD65546-554-specific CTL with the β-cells leads to a reduction in insulin production and the induction of perforin-mediated cell death. Collectively, these findings support a role for the cross-presentation of GAD65 antigen in the priming and enhancement of dominant GAD65-specific CTL responses, which can directly target β-cells that display GAD65 epitopes.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by the activation of autoreactive T cells specific for the insulin-producing pancreatic β-cells, resulting in β-cell destruction and ultimately, the loss of blood glucose homeostasis [1]. The non-obese diabetic (NOD) mouse is an animal model of human T1D. At 4-5 weeks of age, NOD mice spontaneously develop insulitis after which 80-90% of females develop T1D [2-4]. While the NOD mouse has been extensively studied and characterized, the mechanisms that lead to the priming of spontaneous β-cell autoimmunity are still under investigation.
Normally, insulitis and lymphocytic infiltration into the pancreatic islets precedes overt T1D, but such inflammation does not always lead to overt β-cell destruction and disease onset [3, 5]. The events that cause actual β-cell destruction and progression to clinical disease remain unknown, as is the determination of CD4+ or CD8+ T cells as the principal mediators of β-cell death. Previous studies have demonstrated T1D transfer into NOD.scid with CD8+ CTL alone [6, 7], while others have shown disease transfer with CD4+ T cell clones [8, 9]. These findings suggest that both CD4+ and CD8+ T cells are involved in the pathogenesis of T1D [10], and are thus capable of inducing β-cell death. However, pancreatic β-cells express major histocompatibility complex (MHC) class I but lack MHC class II proteins, suggesting that direct cytotoxicity can only be mediated by CD8+ CTL that recognize peptide antigen:MHC class I complexes displayed on β-cells. Moreover, NOD mice deficient in MHC class I or MHC class I associated-β2M are protected from both insulitis and T1D [11-14], demonstrating that MHC class I presentation to CTL is necessary for disease initiation and progression to T1D. In the inflamed islets, diabetogenic CTL populations can induce overt T1D [7], while other self-reactive CTL may contribute via exacerbation of insulitis and/or the activation of T1D-causing T cells [15].
Several studies have shown that CTL are cytotoxic to islet cells [16-18]. CTL can kill target cells via perforin and granzyme B secretion and/or Fas-Fas ligand interactions upon epitope recognition [17, 19, 20]. The primary method of CTL-mediated islet cell death is likely via perforin/granzyme B [21], although CTL may also secrete cytokines such as IFNγ and TNFα that can provide an indirect method for β-cell damage [22]. Islet cells treated with proinflammatory cytokines can upregulate MHC class I and increase their susceptibility to CTL-mediated cytotoxicity [22]. Additionally, treatment with cytokines can lead to increased expression of FasL (CD178), suggesting that Fas-FasL interaction may be a secondary form of CTL-mediated cytotoxicity [22].
Glutamic acid decarboxylase 65kD (GAD65), an enzyme expressed in the pancreatic β-cells, is a candidate autoantigen in the initiation and pathogenesis of T1D [9, 23, 24]. T cell responses to GAD65 are detectable in the spleens of prediabetic NOD mice and in the peripheral blood of humans with T1D prior to the onset of overt hyperglycemia [23, 24], suggesting that GAD65 autoimmunity is important in the initial and later stages of disease. While the CD4+ responses to GAD65 determinants have been well characterized [23-25], less is known about the CTL response to GAD65 [26-29]. Previously, we have reported that peptide 546-554 is a pathogenic CTL-inducing epitope of GAD65 [26, 28]. GAD65546-554-specific CTL are detected in naïve NOD mice, produce inflammatory cytokines IFNγ and TNFα upon culture with cognate antigen [26], and transfer islet inflammation [28] Importantly, subsequent work has confirmed that GAD65-specific CTL, including those specific for p546-554, have diabetogenic potential [29]. Because of challenges to isolating pure β-cells from the pancreas, whole islet cell populations are often used to study CTL-mediated β-cell cytotoxicity. Pure β-cells offer the opportunity to investigate direct β-cell-CTL interactions. In this report, we show direct communication between GAD65546-554-reactive CTL and a pancreatic β-cell line, which led to a loss of insulin production and β-cell death.
Materials and methods
Mice and cells
NOD and NOD.scid mice were purchased from Taconic Farms (Germantown, NY) and Jackson Laboratories (Bar Harbor, ME), respectively, and were housed in the University of Toledo Animal Care Facility. All experiments described in this report were reviewed and approved by the University of Toledo's Institutional Animal Care and Use Committee. NOD NIT-1 insulinoma cells were obtained from Dr. David Serreze (Jackson Laboratories; Bar Harbor, ME). GAD65-expressing NIT-1 cells were created by transfecting NIT-1 cells [30] with pcDNA3.GAD plasmid and subsequent selection with Genetecin (G418). Cloning of G418-resistant cells led to the recovery of several GAD-expressing NIT-1 cell lines, including NIT.GAD.A2. NIT.GAD.A2 cells were cultured in complete media (RPMI supplemented with fetal bovine serum, penicillin/streptomycin, 2-mercapto-ethanol, sodium pyruvate, and nonessential amino acids). HIT-15 insulinoma cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in complete medium, while the B cell lymphoma line M12C3 (Kd) and the 4G4 B cell hybridoma (H-2d/g7) were grown in RPMI supplemented with 10% horse serum.
GAD65546-554-specific CTL lines and clones were generated as previously described [28] following immunization of NOD mice with GAD65546-554 (SYQPLGDKV) (Invitrogen, Carlsbad, CA). NP147-specific CTL were generated by immunization of NOD mice with the Kd-binding peptide, 147-155 (TYQRTRALV), from the nucleoprotein (NP) of influenza. Briefly, mice were immunized subcutaneously with 20-50 μg of peptide in CFA. Ten to 12 days later, single cell suspensions (8×106/ml) from draining lymph nodes were cultured in complete medium with 10 μg/ml of cognate peptide. Recombinant human IL-2 was added the next day (10 ng/ml). Peptide-responsive cells were restimulated every 2 wk with irradiated syngeneic spleen cells pulsed with cognate peptide, and maintained in IL-2-containing medium. Control cells include Th clones specific for GAD65 peptide 530-543 and Th clones specific for myelin oligodendrocyte glycoprotein (MOG) peptide 35-55.
Immunohistochemistry
Following adoptive transfer, pancreas organs from recipients were collected and fixed in 10% buffered formalin and embedded in paraffin. Serial sections, 5-7 μm thick, and 20 μm apart were placed on glass slides. Prior to immunostaining the pancreatic sections were deparaffinized with SafeClear (Fisher Scientific). NIT.GAD.A2 cells were grown overnight on glass cover slips and fixed with 4% paraformaldehyde. Both the pancreatic tissue and β-cell slides were treated with saponin and blocked with 2% horse serum prior to the addition of antibodies. GAD65-specific monoclonal antibodies, GAD-1 (ATCC) and GAD-6 (a gift from Dr. Dan Zekzer), were purified from the supernatants of the corresponding mouse B cell hybridomas by Protein-G column chromatography. The human/mouse/bovine insulin-specific antibody was purchased from R&D Systems (Minneapolis, MN), goat-anti-mouse IgG (H&L)-biotin was purchased from Southern Biotech (Birmingham, AL), while StrepAvidin-Alexa Fluor-488 and Avidin-HRP were purchased from Invitrogen and Vector Labs (Burlingame, CA), respectively. Primary antibodies were added and incubated overnight at 4°C, while secondary reagents were added for 1 hr each at room temperature. For sections treated with Avidin-HRP, a peroxidase staining kit (Vector Labs) was used to visualize enzyme deposition.
Cytotoxicity assay
An MTT assay was used to determine CTL-induced loss of viability among NIT.GAD.A2 cells (1×105 cells/well) cultured with GAD65546-554-specific or NP147-specific CTL (1×105 cells/well) in 96-well flat-bottom plates. Forty-eight hr after co-culture, triplicate wells were gently washed to remove nonadherent cells and debris before the addition of a MTT solution (0.5mg/ml) (Sigma Aldrich, St. Louis, MO) and incubated for 1 hr at 37°C. Finally, 10% SDS in 0.01N HCl was added to the wells overnight at 37°C, after which the absorbance of each well was determined at 595 nm using a Bio-Rad microplate reader. Viability was calculated as follows:
Absorbance of experimental wells (CTL + target cells)/Absorbance of control wells (target cells only)
To determine CD95 (Fas)-mediated loss of cell viability, NIT.GAD.A2 or M12C3 target cells were plated (2×105 cells/well) with soluble anti-Fas monoclonal Ab (0.5 μg/ml; Clone Jo2, BD Biosciences) and crosslinked with protein G (1.0 g/ml) (Sigma) for 48 hr at 37°C. The MTT assay was performed as above to determine cell viability.
To evaluate the metabolic activity of NIT.GAD.A2 cells, an insulin-specific radioimmunoassay (Linco Research, St Charles, MO) was used to determine the hormone level in supernatants from co-culture experiments. Briefly, equal numbers of NIT.GAD.A2 cells (1×105 cells/well) and antigen-specific CTL were placed in complete medium, in 96-well flat bottom plates. Supernatants from replicate wells were collected after 48 hr and diluted with buffer as per the manufacturer’s instructions, tested in triplicate, and subsequently compared to a standard curve of purified insulin.
ELISPOT assay
ELISPOTs were performed as previously described [28]. Briefly, Multiscreen HA filtration plates (Millipore, Billerica, MA) were coated with anti-mouse IFNγ overnight. After blocking non-specific sites, 1-10×104 antigen-specific CTL were co-cultured with 3×105 NIT.GAD.A2, NIT, HIT-15, or M12C3 cells in complete medium for 24 hr. The plates were washed and biotinylated anti-mouse IFNγ was added for at least 6 hr, followed by the addition of avidin-HRP for 1 hr. IFNγ-secreting spots were detected by adding chromogenic substrate, and then enumerated using a stereo microscope.
Flow cytometry
To evaluate perforin expression, the CTL were fixed with 4% paraformaldehyde, resuspended in permeabilization buffer (1% saponin and 0.05% sodium azide in PBS), stained with PE-conjugated anti-Perforin or Rat Ig-PE (eBioscience, San Diego, CA), and analyzed by flow cytometry. CTL were identified by size and granularity (forward and side scatter). To detect IFNγ production in CTL after culture with NIT.GAD.A2 cells, GAD65546-554-specific CTL were cultured with 4G4 cells, pulsed with p546-554 or buffer, or NIT.GAD.A2 cells in the presence of Brefeldin A (5 μg/ml) (Sigma Adrich; St Louis, MO) for 6 hr at 37°C. The cells were washed, fixed with 4% paraformaldehyde, and resuspended in permeabilization buffer. Next, 0.4 μg of PE-conjugated anti-IFNγ (BD Biosciences, San Diego, CA) or isotope control (Ig-PE) was added for an additional 30 min at 4°C, after which the cells were washed and analyzed by flow cytometry. CTL were identified by size and granularity (forward and side scatter).
To evaluate CD95 expression on NIT.GAD.A2, the cells were trypsinized, washed, and stained stepwise with biotinylated anti-CD95 (eBiosciences, San Diego, CA) and StrepAvidin-AlexaFluor 488, and then analyzed by flow cytometry. Controls were stained with isotype control plus secondary or secondary reagent alone.
Results
GAD65546-554 -specific CTL lines and clones induce insulitis and localized loss of β-cell insulin production in NOD.scid recipients
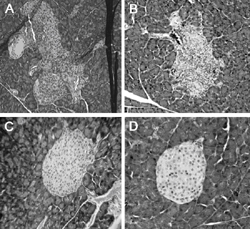
GAD65 p546-554-specific T cells are detectable in the spleens of prediabetic NOD mice. As we previously reported, adoptively transferred GAD546-554-specific CTL cause insulitis in NOD.scid recipient mice [28]. The islets of recipient mice showed characteristic lymphocyte infiltration and disruption of islet architecture (Figures 1A-B); while the islets of mice that received an equal number of T cells from a CTL line specific for GAD268-78 (Figure 1C), or saline (Figure 1D), remained healthy and showed no signs of lymphocytic infiltration. Additionally, GAD65546-554-specific CTL induced localized islet-specific loss of insulin production (Figures 2A-C and 2E), which was not obvious in healthy NOD.scid (Figure 2D) or the GAD268 controls (data not shown).
 |
 |
Figure 1. Adoptive transfer of GAD65546-554-specific CTL induces insulitis in NOD.scid recipients. Recently activated GAD65546-554-specific CTL (1×107) were suspended in saline and injected into the peritoneal cavity of NOD.scid mice. Eleven days later the mice were sacrificed, and the pancreata were formalin-fixed, paraffin-embedded, sectioned, and stained with hematoxylin and eosin. Representative sections were photographed and evaluated for insulitis. A: islet from the recipient of CTL line GAD 546.14 (10X). B: islet from the recipient of CTL line GAD 546.16 (20X). C: islet from the recipient of CTL line GAD 268.11 (20X). D: islet from saline-treated NOD.scid (40X). |
|
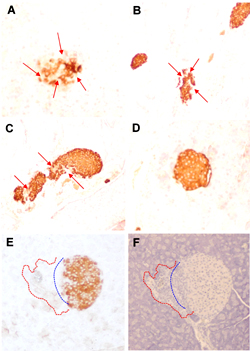
Figures 2E and 2F reveal a relationship between lymphocyte infiltration and the loss of insulin staining (insulin or hematoxylin staining of adjacent sections). The H&E sections showed that the vast majority of the infiltrating cells in disrupted islets were lymphocytes. Given the purity of the CTL lines and clones that were transferred (>95% CD8+ and TCR+ at the time of transfer) and the lack of host lymphocytes in NOD.scid recipients, it is clear that the islet infiltrating cells contained the GAD546-specific CTL. The infiltrating cells were also associated with disruption of normal islet architecture (Figures 2E and 2F). However, despite the obvious damage to numerous islets, we did not observe overt diabetes in the NOD.scid recipients. Our GAD65 CTL may be important in the early phases of insulitis and β-cell damage, which leads to the recruitment of Th cells and CTL of other specificities that may be needed to progress to diabetes in NOD.scid mice. Our findings indicate that infiltrating GAD-specific CTL can directly interact with insulin-producing β-cells, disrupt islet structure, and induce β-cell damage.
 |
|
Figure 2. Adoptive transfer of GAD65546-554-specific CTL leads to a loss of insulin production in vivo. Recently activated GAD65546-554-specific CTL (1×107) were i.p. injected into NOD.scid mice. Eleven days later, the mice were sacrificed and the pancreata were fixed, embedded, sectioned, and immunostained to reveal sites of insulin production. Representative sections were photographed. A: islet from recipient of CTL clone GAD 546.14.46. B: islet from recipient of CTL line GAD 546.16. C: islet from recipient of CTL line GAD 546.14. D: healthy islet of a naïve NOD.scid. Red arrows indicate areas of localized insulin loss. The two bottom panels (E and F) are adjacent sections of a representative islet from a recipient of CTL line GAD 546.16. E: immunohistochemical staining with anti-insulin. F: hematoxylin alone. The dashed blue line outlines the theoretical extrapolation of the islets normal border, while the dashed red line encircles an area showing lymphocyte infiltration that disrupts islet architecture. |
|
GAD65-transfected NIT β-cell insulinoma expresses GAD65 and insulin at detectable levels
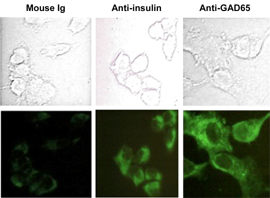
A key to unraveling autoimmune diabetes requires an understanding of the interaction between immune cells and insulin-producing β-cells. Investigations of immune-mediated β-cell death often involve the use of primary islet cells, which contain a mixture of α-, β-, and δ-cell populations. The isolation of purified β-cells from islet cell preparations requires tedious mechanical and enzymatic manipulations. Furthermore, typical cytotoxicity assays can be problematic when evaluating T cell activity because β-cells are sensitive to 51Cr exposure and are poor loaders of 3H thymidine (unpublished data). Because of the fastidious nature of purified islet β-cells, we chose NIT-1 β-cell insulinoma cells to aid in our investigation of β-cell-CTL specific interactions. NIT-1 is a well-established NOD β-cell line [30] that produces insulin in vitro, but has lost the ability to express GAD65 at detectable levels, which can also be difficult to detect in preparations of mouse islets [31]. To ensure stable GAD65 expression, we transfected the NIT-1 β-cell line with a GAD65-expressing plasmid and subcloned several stable cell lines. One of the resulting lines, NIT.GAD.A2, expresses both GAD65 and insulin at levels detectable by immunofluorescence (Figure 3).
 |
|
Figure 3. NIT.GAD.A2 cells express insulin and GAD65. NIT.GAD.A2 cells were grown overnight on glass cover slips, fixed, permeabilized, and incubated with mouse IgG, anti-insulin, or anti-GAD65. The slides were then treated stepwise with biotinylated goat-anti-mouse IgG and strepavidin-AlexaFluor 488. The top panels depict phase-contrast brightfield images, while the bottom panels are fluorescent images of the same cells captured under UV light. |
|
GAD65546-554-specific CTL kill NIT.GAD.A2 in an epitope-restricted manner
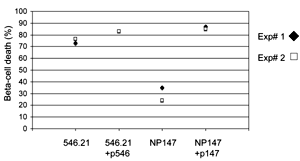
We used an MTT assay to determine whether GAD65546-554-specific CTL could directly cause a loss of viability in NIT.GAD.A2 β-cells. Viable NIT.GAD.A2 are adherent, so the gentle washing needed for analysis does not disturb a viable monolayer. The viability of NIT.GAD.A2 β-cells was reduced by more than 70% when co-cultured with the GAD65546-554-specific CTL clone, 546.21 (Figure 4), but similar responses were not seen when the β-cells were cultured with the influenza-specific CTL-line, NP147 (Figure 4). In some experiments, we observed a slight loss of NIT.GAD.A2 viability when NP147 CTL were added (Figure 4). However, the level of killing never reached the magnitude observed with GAD-specific CTL, and was attributed to non-specific interactions since dramatic increases in cell death occurred when NIT.GAD.A2 cells were prepulsed with peptide NP147 (Figure 4). In contrast to this result, prepulsing with GAD546-554 peptide did not increase the killing observed with the GAD-specific CTL (Figure 4).
 |
|
Figure 4. GAD65546-554-specific CTL can kill GAD-expressing β-cells. NIT.GAD.A2 cells (1×105 c/w) were incubated with an equal number of GAD-specific CTL line 546.21 or the control CTL line NP147 (specific for residues 147-155 of NP molecule of influenza), for 48 hr at 37°C in 96-well plates. Positive controls included NIT.GAD.A2 cells prepulsed with cognate peptide (10 μg/ml of p546-554 or p147-155). The plates were washed to remove CTL and debris before cell viability was measured in each well. Cell viability (% cell death) was calculated as described in the materials and methods section. The filled circles and open squares reflect results from two independent experiments, but they are representative of multiple experiments with a number of different GAD65546-554-specific CTL lines and clones. The mean values were calculated from triplicate wells. |
|
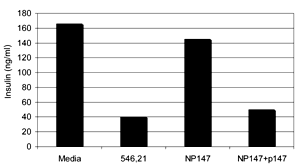
In addition to cell death, we found that co-culture with GAD65546-554-specific CTL reduced the production of insulin by NIT.GAD.A2 by greater than 75% (Figure 5).
 |
|
Figure 5. GAD65546-554-specific CTL diminish insulin production in vitro. NIT.GAD.A2 (1×105 c/w) were incubated with an equal number of GAD-specific, 546.21, or control CTL NP147 for 48 hr at 37°C in 96-well plates. NP147 CTL were also incubated with cognate peptide-pulsed NIT.GAD.A2 cells (NP147+peptide). Culture supernatants were collected and analyzed in an insulin-specific RIA. Mean values were calculated from triplicate wells. |
|
GAD65546-554-specific CTL kill NIT.GAD.A2 via a FasL-independent mechanism
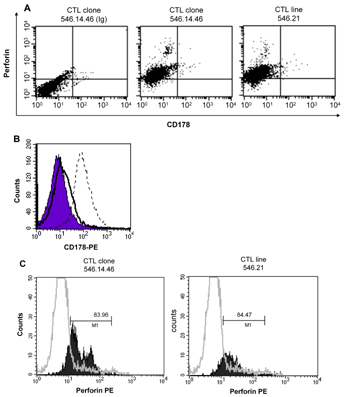
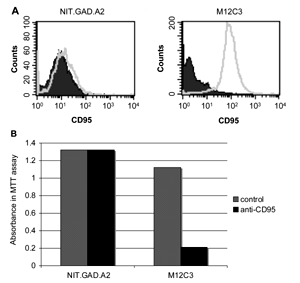
To determine the mechanisms of cytolytic activity of GAD65546-554-reactive CTL against NIT.GAD.A2, we evaluated the expression of the death-related molecules CD178 and perforin on our CTL. We found that CTL clone 546.14.46 and CTL line 546.21 each expressed perforin (Figures 6A and 6C). However, CD178 was undetectable on both lines (Figures 6A and 6B). Additionally, in spite of their susceptibility to the GAD-specific CTL, we could not detect CD95 expression on NIT.GAD.A2 β-cells (Figure 7A), suggesting that Fas-FasL interactions were not centrally involved in GAD65546-554-specific CTL mediated β-cell cytotoxicity. We readily detected CD95 on M12C3 cells (Figure 7A), which were also killed by the GAD-specific CTL when pulsed with cognate peptide. To confirm that NIT.GAD.A2 were not killed in a Fas-FasL-dependent manner, we performed a cytotoxicity assay with soluble anti-CD95 monoclonal antibody, which induced apoptosis in CD95-expressing cells. While NIT.GAD.A2 were completely refractive to apoptosis when cultured with anti-CD95, similar treatment of the M12C3 cells induced a greater than 80% loss of viability (Figure 7B). Collectively, these data demonstrated that the GAD65546-554-specific CTL could kill β-cells in CD178-independent manner.
 |
|
Figure 6. GAD65546-554-specific CTL express perforin but not CD178. GAD-specific CTL clone 546.14.46 and CTL line 546.21, and the MOG-specific (myelin oligodendrocyte glycoprotein) Th cell clone SC5 were stained with isotype control antibodies (IgG-PE), or PE-conjugated anti-perforin or biotinylated anti-CD178 (FasL) plus avidin-Alexa Fluor 488, as described in the materials and methods section. The cells were analyzed by flow cytometry. A: cells within the lymphocyte gate are displayed as dot plots. B: CTL 546.14.46 was stained with anti-CD178 (solid line) or isotype control (filled histogram). The Th cell clone GAD530.1 was also stained with isotype control (data not shown) or anti-CD178 (broken line) as a positive control for CD178 expression. C: Data from a second experiment are displayed as histograms. The MOG-specific Th cell clone is shown as the unfilled histogram. The values in the histogram reveal the percentage of cells that were positive compared to isotype control. These results are representative of two experiments. |
|
 |
|
Figure 7. GAD CTL kill NIT.GAD.A2 cells via a CD95-independent mechanism. A: NIT.GAD.A2 or M12 C3.PIA cells (1.5×105) were stained with isotype control (filled histogram) or biotinylated-anti-CD95 (Fas) (unfilled histogram) followed by StrepAvidin-AlexaFluor 488. B: NIT.GAD.A2 cells or M12C3.PIA were plated (2×105 c/w) with soluble anti-Fas monoclonal Ab (0.5 μg/ml) and protein G (1.0 μg/ml). After 48 hr at 37°C, an MTT assay was used to evaluate cell viability. The mean values and SD were calculated from triplicate wells and the results are representative of two experiments. |
|
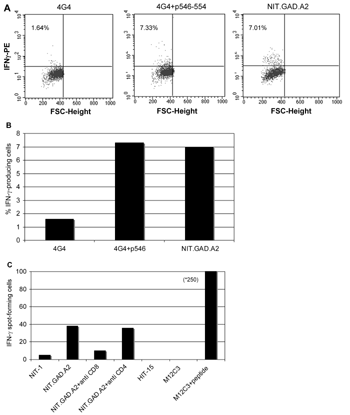
Cytokine production is an additional indicator of CTL activation and recognition of cognate epitope. GAD65546-554-specific CTL produced IFNγ when cultured with NIT.GAD.A2, as shown by intracellular cytokine staining (Figures 8A and 8B) and ELISPOT assay (Figure 8C). Interestingly, compared to the M12C3 cells, the β-cell line was relatively poor at inducing cytokine release (Figure 8C), which we could not compensate for by adding more β-cells to the culture. Two-thirds fewer peptide-pulsed M12C3 cells yielded 5-fold more spots than the β-cells (Figure 8C). Nevertheless, the results demonstrated β-cell display of the CTL epitope and subsequent T cell activation. Therefore, NIT.GAD.A2 cells were capable of processing 546-554 from the GAD65 molecule. However, despite their ability to produce GAD65 (detected by immunofluorescence, data not shown), hamster-derived HIT-15 insulinomas were incapable of inducing IFNγ release from the GAD-reactive CTL (Figure 8), which confirmed that the CTL are both GAD65-specific and Kd-restricted [28], and this crosspresentation was not contributing to our assay results.
 |
|
Figure 8. GAD546-554-specific CTL produce IFNγ in the presence of GAD-expressing β-cells. CTL clone 546.14.74 CTL (5×104 c/w) and 4G4 B cell hybriodmas (1×105 c/w), prepulsed with cognate peptide p546-554 or saline, or NIT.GAD.A2 were co-cultured in the presence of BFA for 6 hr at 37°C. The cells were fixed, permeabilized, and stained with PE-conjugated (A) anti-IFNγ or isotype control (data not shown). The figure shown is representative of two independent experiments. Figure B is a graphical representation of the FACS dot plots in A. C: ELISPOT analysis for IFNγ production. CTL (5×104 c/w) were co-cultured with 3×105 NIT.GAD.A2, NIT-1, or HIT-15 insulinomas (3×105 c/w) in 96 well ELISPOT plates and then processed as described in the materials and methods section. As positive control, 1×103 CTL were cultured with 1×105 M12C3 cells, pulsed with or without cognate peptide. The data represent mean spot-forming cells of triplicate wells. The standard deviations were less than 5%. |
|
Discussion
Much of our knowledge about T cell to β-cell interactions emanates from previous investigations of self-antigen-specific CTL-mediated β-cell death performed with islet cell populations or investigations utilizing monoclonal T cells specific for a non-β-cell antigen expressed as a transgene in islet cells [19]. Previously, we have shown that the peptide p546-554 contains a dominant Kd-restricted determinant of GAD65 that induces CTL capable of driving insulitis in NOD mice [28]. We now report evidence of direct interaction between GAD65546-554-specific CTL and a β-cell line. Importantly, we found that GAD65-specific CTL, and not NP147-specific CTL, mediated intense β-cell cytotoxicity in the absence of professional APC or phagocytes. The lack of comparable NIT.GAD.A2 β-cell death with the irrelevant CTL demonstrates that, in spite of their sensitivity to the deleterious effects inflammation, the direct presentation of MHC:peptide ligands to CTL accounts for the majority of the β-cell death we observed in co-culture experiments.
Previous studies suggest that perforin/granzyme B secretion is the primary method of CTL-induced β-cell cytotoxicity [32] and that Fas-FasL-dependent killing is likely to occur in the absence of perforin [17, 19, 20]. Collectively, our data supports this conclusion as GAD65546-554-specific CTL kill NIT.GAD.A2 β-cells in a FasL-independent, and likely perforin-dependent manner. While combinations of proinflammatory cytokines can induce the expression of CD95 on NIT cells [33], we did not detect increased CTL-mediated cytotoxicity of NIT.GAD.A2 in the presence of IFNγ and TNFα (unpublished data).
In addition to direct target cell cytolysis, we report a loss of insulin production in β-cells co-cultured with GAD65546-554-specific CTL, but not with NP147, suggesting that insulin loss is related to antigen-specific presentation by the β-cells and is due to direct β-cell damage and not CTL absorption of secreted insulin. In vivo, we observed localized insulin loss in the islets of NOD.scid mice that received GAD65546-554-specific CTL, and the loss coincided with islet infiltration by lymphocytes. Further investigation is required to define the kinetics of insulin loss and clarify whether substantial loss of the metabolic function occurs prior to cell death.
Additionally, NIT.GAD.A2 cells induced low level CTL secretion of IFNγ. IFNγ signaling through the Jak-STAT pathway is prolonged in β-cells [34], and may upregulate MHC class I, rendering them more susceptible to CTL-mediated cytolysis [35]. Therefore, in the inflamed islet, IFNγ is implicated in both direct and indirect β-cell damage, as localized tissue damage cytokine release by CTL may promote pancreatic inflammation and recruit other immune mediators including APC and Th cells.
Our demonstration of direct β-cell cytotoxicity and CTL activation highlights the ability of β-cells to naturally and preferentially process and present CTL-inducing GAD65 epitopes that have diabetogenic potential [29]. Importantly, p546-554 is processed and presented in GAD65-transfected B cells, as well as macrophages/dendritic cells and β-cells that can capture or express GAD65 in vivo, respectively [28]. This finding shows that the determinant is intrinsically accessible and readily excised from GAD65 by both β-cells and professional APC. Therefore, cross-presentation of β-cell antigens released following tissue damage by infectious agents [36], age-related tissue remodeling [37], and/or normal cellular turnover may be a significant mechanism of priming autoreactive CD8+ T cells.
In the pancreas, activated GAD65-specific CTL can exclusively initiate an immune response via direct TCR-MHC I interactions with β-cells, a feature not possessed by Th cells. The mechanisms by which Th cells mediate T1D remains unclear; however, such cells may secrete inflammatory cytokines or other soluble mediators that preferentially damage surrounding β-cells, but leave other islet cells unscathed. Speculations on the precise contribution of GAD65 to T1D pathogenesis have been controversial, and its role in islet inflammation also remains elusive. We and others have found GAD65546-554-specific CTL in naïve NOD mice [26, 29]. Treatment of NOD mice with GAD65 protein or peptides can induce tolerance and protection from T1D, or enhance the progression of the disease [9, 29]. GAD65 autoimmunity may play an even greater role in human T1D pathogenesis [38], as human β-cells synthesizes far more GAD65 than murine β-cells.
Clearly, not all GAD65 epitopes are immunogenic and it is likely that only a limited number are able to recruit T cells that influence the course of T1D. Moreover, some epitopes with diabetogenic potential may not be readily processed in vivo by the appropriate cells. The processing and presentation of relevant islet cell antigens by β-cells may contribute significantly to the susceptibility and progression of T1D. The ability to promptly identify these critical epitopes will be important in our efforts to design therapeutic interventions that can arrest β-cell-specific inflammatory mechanisms.
Conflict of interest statement: The authors declare that they have no competing conflict of interests with respect to financial or other issues.
References
- Eisenbarth G. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 1986. 314(21):1360-1368. [DOD]
- Serreze DV, Leiter EH. Genetic and pathogenic basis of autoimmune diabetes in NOD mice. Curr Opin Immunol 1994. 6(6):900-906. [DOD] [CrossRef]
- Wicker LS, Todd JA, Peterson LB. Genetic control of autoimmune diabetes in the NOD mouse. Annu Rev Immunol 1995. 13:179-200. [DOD] [CrossRef]
- Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 1997. 7(6):727-738. [DOD] [CrossRef]
- Miyazaki A, Hanafusa T, Yamada K, Miyagawa J, Fujino-Kurihara H, Nakajima H, Nonaka K, Tarui S. Predominance of T lymphocytes in pancreatic islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: a longitudinal study. Clin Exp Immunol 1985. 60(3):622-630. [DOD]
- Graser RT, DiLorenzo TP, Wang F, Christianson GJ, Chapman HD, Roopenian DC, Nathenson SG, Serreze DV. Identification of a CD8 T cell that can independently mediate autoimmune diabetes development in the complete absence of CD4 T cell helper functions. J Immunol 2000. 164(7):3913-3918. [DOD]
- Wong FS, Visintin I, Wen L, Flavell RA, Janeway CA Jr. CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J Exp Med 1996. 183(1):67-76. [DOD] [CrossRef]
- Peterson JD, Pike B, McDuffie M, Haskins K. Islet-specific T cell clones transfer diabetes to nonobese diabetic (NOD) F1 mice. J Immunol 1994. 153(6):2800-2806. [DOD]
- Zekzer D, Wong FS, Ayalon O, Millet I, Altieri M, Shintani S, Solimena M, Sherwin RS. GAD-reactive CD4+ Th1 cells induce diabetes in NOD/SCID mice. J Clin Invest 1998. 101(1):68-73. [DOD] [CrossRef]
- Miller BJ, Appel MC, O'Neil JJ, Wicker LS. oth the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol 1988. 140(1):52-58. [DOD]
- Katz J, Benoist C, Mathis D. Major histocompatibility complex class I molecules are required for the development of insulitis in non-obese diabetic mice. Eur J Immunol 1993. 23(12):3358-3360. [DOD] [CrossRef]
- Sumida T, Furukawa M, Sakamoto A, Namekawa T, Maeda T, Zijlstra M, Iwamoto I, Koike T, Yoshida S, Tomioka H, et al. Prevention of insulitis and diabetes in beta 2-microglobulin-deficient non-obese diabetic mice. Int Immunol 1994. 6(9):1445-1449. [DOD] [CrossRef]
- Serreze DV, Leiter EH, Christianson GJ, Greiner D, Roopenian DC. Major histocompatibility complex class I-deficient NOD-B2mnull mice are diabetes and insulitis resistant. Diabetes 1994. 43(3):505-509. [DOD] [CrossRef]
- Wicker LS, Leiter EH, Todd JA, Renjilian RJ, Peterson E, Fischer PA, Podolin PL, Zijlstra M, Jaenisch R, Peterson LB. Beta 2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes 1994. 43(3):500-504. [DOD] [CrossRef]
- Serreze DV, Chapman HD, Varnum DS, Gerling I, Leiter EH, Shultz LD. Initiation of autoimmune diabetes in NOD/Lt mice is MHC class I-dependent. J Immunol 1997. 158(8):3978-3986. [DOD]
- Nagata M, Yokono K, Hayakawa M, Kawase Y, Hatamori N, Ogawa W, Yonezawa K, Shii K, Baba S. Destruction of pancreatic islet cells by cytotoxic T lymphocytes in nonobese diabetic mice. J Immunol 1989. 143(4):1155-1162. [DOD]
- Dudek NL, Thomas HE, Mariana L, Sutherland RM, Allison J, Estella E, Angstetra E, Trapani JA, Santamaria P, Lew AM, et al. Cytotoxic T-cells from T-cell receptor transgenic NOD8.3 mice destroy beta-cells via the perforin and Fas pathways. Diabetes 2006. 55(9):2412-2418. [DOD] [CrossRef]
- Campbell PD, Estella E, Dudek NL, Jhala G, Thomas HE, Kay TW, Mannering SI. Cytotoxic T-lymphocyte-mediated killing of human pancreatic islet cells in vitro. Hum Immunol 2008. 69(9):543-551. [DOD] [CrossRef]
- McKenzie MD, Dudek NL, Mariana L, Chong MM, Trapani JA, Kay TW, Thomas HE. Perforin and Fas induced by IFNgamma and TNFalpha mediate beta cell death by OT-I CTL. Int Immunol 2006. 18(6):837-846. [DOD] [CrossRef]
- Sutton VR, Estella E, Li C, Chen M, Thomas HE, Kay TW, Trapani JA. A critical role for granzyme B, in addition to perforin and TNFalpha, in alloreactive CTL-induced mouse pancreatic beta cell death. Transplantation 2006. 81(2):146-154. [DOD] [CrossRef]
- Kreuwel HT, Morgan DJ, Krahl T, Ko A, Sarvetnick N, Sherman LA. Comparing the relative role of perforin/granzyme versus Fas/Fas ligand cytotoxic pathways in CD8+ T cell-mediated insulin-dependent diabetes mellitus. J Immunol 1999. 163(8):4335-4341. [DOD]
- Rabinovitch A. An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Diabetes Metab Rev 1998. 14(2):129-151. [DOD] [CrossRef]
- Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GS, Robinson P, Atkinson MA, Sercarz EE, Tobin AJ, Lehmann PV. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature 1993. 366(6450):69-72. [DOD] [CrossRef]
- Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature 1993. 366(6450):72-75. [DOD] [CrossRef]
- Chao CC, McDevitt HO. Identification of immunogenic epitopes of GAD65 presented by Ag7 in non-obese diabetic mice. Immunogenetics 1997. 46(1):29-34. [DOD] [CrossRef]
- Quinn A, McInerney MF, Sercarz EE. MHC class I-restricted determinants on the glutamic acid decarboxylase 65 molecule induce spontaneous CTL activity. J Immunol 2001. 167(3):1748-1757. [DOD]
- Ejrnaes M, Videbaek N, Christen U, Cooke A, Michelsen BK, von Herrath M. Different diabetogenic potential of autoaggressive CD8+ clones associated with IFN-gamma-inducible protein 10 (CXC chemokine ligand 10) production but not cytokine expression, cytolytic activity, or homing characteristics. J Immunol 2005. 174(5):2746-2755. [DOD]
- Busick RY, Aguilera C, Quinn A. Dominant CTL-inducing epitopes on GAD65 are adjacent to or overlap with dominant Th-inducing epitopes. Clin Immunol 2007. 3:298-311. [DOD] [CrossRef]
- Severe S, Gauvrit A, Vu AT, Bach JM. CD8+ T lymphocytes specific for glutamic acid decarboxylase 90-98 epitope mediate diabetes in NOD SCID mouse. Mol Immunol 2007. 44(11):2950-2960. [DOD] [CrossRef]
- Hamaguchi K, Gaskins HR, Leiter EH. NIT-1, a pancreatic beta-cell line established from a transgenic NOD/Lt mouse. Diabetes 1991. 40(7):842-849. [DOD] [CrossRef]
- Faulkner-Jones BE, Cram DS, Kun J, Harrison LC. Localization and quantitation of expression of two glutamate decarboxylase genes in pancreatic beta-cells and other peripheral tissues of mouse and rat. Endocrinology 1993. 133(6):2962-2972. [DOD] [CrossRef]
- Thomas HE, Darwiche R, Corbett JA, Kay TW. Evidence that beta cell death in the nonobese diabetic mouse is Fas independent. J Immunol 1999. 163(3):1562-1569. [DOD]
- Augstein P, Dunger A, Salzsieder C, Heinke P, Kubernath R, Bahr J, Fischer U, Rettig R, Salzsieder E. Cell surface trafficking of Fas in NIT-1 cells and dissection of surface and total Fas expression. Biochem Biophys Res Commun 2002. 290(1):443-451. [DOD] [CrossRef]
- Chong MM, Thomas HE, Kay TW. Gamma-interferon signaling in pancreatic beta-cells is persistent but can be terminated by overexpression of suppressor of cytokine signaling-1. Diabetes 2001. 50(12):2744-2751. [DOD] [CrossRef]
- Thomas HE, Parker JL, Schreiber RD, Kay TW. IFN-gamma action on pancreatic beta cells causes class I MHC upregulation but not diabetes. J Clin Invest 1998. 102(6):1249-1257. [DOD] [CrossRef]
- Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med 1998. 4(7):781-785. [DOD] [CrossRef]
- Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 1995. 44(3):249-256. [DOD] [CrossRef]
- Blancou P, Mallone R, Martinuzzi E, Severe S, Pogu S, Novelli G, Bruno G, Charbonnel B, Dolz M, Chaillous L, van Endert P, Bach JM. Immunization of HLA class I transgenic mice identifies autoantigenic epitopes eliciting dominant responses in type 1 diabetes patients. J Immunol 2007. 178(11):7458-7466. [DOD]
This article has been cited by other articles:
|


)
)
)
)
)
)
)
)


