Editorial
| Rev Diabet Stud,
2009,
6(2):76-80 |
DOI 10.1900/RDS.2009.6.76 |
Breakthrough in Diabetes Therapy ... Just Around the Corner?
William A. Rudert, Massimo Trucco
Division of Immunogenetics, Department of Pediatrics, Children´s Hospital of Pittsburgh Rangos Research Center, Pittsburgh, PA 15224, USA
Address correspondence to: Massimo Trucco, e-mail: mnt@pitt.edu
Manuscript submitted August 1, 2009; resubmitted September 7, 2009; accepted September 9, 2009.
Keywords: type 1 diabetes, beta-cell regeneration, T lymphocyte, antibody, autoimmunity, aCD3, aCD4, regulatory T cell, B cell, rituximab, transplantation
Abstract
Inspired by the articles presented in this issue of The Review of Diabetic Studies, we considered it useful to summarize the latest achievements and current challenges we face in the search for a cure of type 1 diabetes. In this editorial article, we took into account how the research landscape has changed in only a few years. While modern lifestyles impose new concerns, now we have a better knowledge of the various aspects of the disease that can be used to treat our young patients with more appropriate approaches, thereby eliminating old and obsolete prejudices.
Introduction
Science is like the waves of the sea constantly advancing and retreating. It takes three steps forward, and two steps back, another three forward and two back. Progress is built on clever intuitions, limited by glorious mistakes; expands based on weird ideas frequently proven wrong by meticulous controls; results out of the hard work of a group of enthusiastic scientists, confirmed or rebutted by the work of another competing group. Some issues remain important, some become less important or disappear with time, some are rediscovered years later under a different light.
Barriers to progress into diabetes research
After many years of discussion, the definition of type 1 diabetes has come to a consensus. This is a syndrome characterized by an onset most frequently presenting in young patients, which is allegedly the result of an autoimmune process that eventually brings about the destruction of the majority of the insulin producing cells in the pancreas, requiring daily administrations of recombinant insulin. In contrast, type 2 diabetes is another syndrome, most frequently seen in obese adults, and is a consequence of the development over time of a target cell resistance to the action of the natively produced insulin. Now this distinction is becoming unclear again since an increased incidence of sedentary lifestyles, synergized with excessive calorific intake, has led to an increase in the frequency of obese children becoming type 1 diabetics [1]. In their review presented in this issue, Coppieters and von Herrath propose a "continuous spectrum", with type 1 and type 2 diabetes positioned at opposite ends. This may well portray quite faithfully the situation we are facing today in our diabetes clinics [2].
Defects in the immune system of type 1 diabetes patients
The once conceptually rejected "suppressor" cells have returned as "regulatory" T (Treg) cells, to complete (or confuse) the immunologic picture of the disease. Physiologically, a pool of bone marrow-derived, immature cells passes through the thymus. Once in the thymus, these cells undergo a receptor allelic exclusion process that generates single positive (SP) thymocytes.
The Ets1 gene, the v-ets erythroblastoid virus E26 oncogene homolog 1, is initially expressed on days 17-18 of murine embryonic development, when SP thymocytes begin to accumulate in large numbers in the thymus. The non-obese diabetic (NOD) mouse is a well-characterized model for type 1 diabetes sharing several of the characteristics of Ets1-deficient targeted mutant mice: defects in T cell receptor (TCR) allelic exclusion and deficiency of natural killer (NK) and NKT (a sub-population that phenotypically stands in between T and NK cells) cell phenotypes [3]. SP thymocytes mature after they are positively and negatively selected through their TCR interaction with fragments of proteins also present in our bodies (self-peptides) and expressed by thymic epithelial cells (TECs). Indeed, the epithelial thymus is now known to express a large array of self-antigens including insulin. All of these are normally produced by cells targeted in a number of autoimmune disorders, one of which is type 1 diabetes.
Human leukocyte antigens (HLA), the human major histocompatibility complex molecules, anchored in the membrane of TECs, display HLA/self-peptide complexes for interaction with the receptor present on those immature T cells passing by. A cell that interacts strongly with the HLA/self-peptide complex dies in the thymus and is thus eliminated, i.e., negatively selected. Also, cells that interact poorly with the complex do not proliferate, become unable to function (i.e., anergic), and are eventually lost. The cells between these two extremes proliferate modestly, survive (i.e., are positively selected), and emerge from the thymus to circulate in the periphery as naïve T cells. HLA molecules, which allow a suboptimal lodging of the antigenic peptide, promote diabetes susceptibility since, under these circumstances, autoreactive T cells may escape negative selection and populate the periphery. However, even individuals with HLA alleles able to provide resistance to the disease may develop the disease, because of a genetically determined, insufficient expression of the self-antigen by the TECs. Once in the circulation, the potentially autoreative, naïve T cells may be activated, even aspecifically as a consequence of environmental stimuli. After finding their target, they begin killing beta-cells, which creates the basis for autoimmunity [4].
Furthermore, in the periphery, T cells that are ready to engage foreign antigens when presented by circulating antigen-presenting cells (APC), can also respond to "ignored" antigens (i.e., those self-antigens not previously exposed to the immune system). This kind of response is amplified by dendritic cells (DC), which are extremely powerful APC, able to present these "new" antigens to naïve T cells through their HLA molecules. The epitope spreading phenomenon (i.e., this expansion of the set of recognized self-antigens) observed in islet inflammation is due to T cells, both CD4- and CD8-positive [5], which were generated in the thymus early in ontogeny but respond to antigens exposed during islet destruction. The continuous presentation of old and new antigens, collected by DC from the newly destroyed beta-cells, to naïve T cells in the pancreatic lymph nodes creates a pathological vicious circle. The newly activated T cells (effector T cells) eventually circulate back to the pancreas to kill other beta-cells, involving at every circle, a larger set of different self-antigens. Treg cells, also matured in the thymus, are committed to fine tuning the tolerogenic apparatus of a healthy immune system. When the level of Tregs is not sufficient to limit the function of effector T cells, type 1 diabetes may slowly develop.
New approaches for intervention
T and B cells
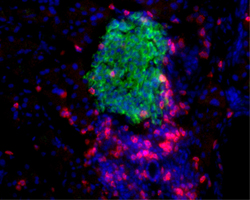
The "siege" of pancreatic islets (i.e., peri-insulitis) (Figure 1) can be considered the result of the interplay between two kinds of cells, effector T cells and regulatory T cells. These cells are supported and complemented in their opposite efforts by many other cells, each with different characteristics and duties in the context of the immune reaction: helper T cells, macrophages, NK, NKT cells and B lymphocytes. Although allelic variation of Ets1 does not seem to contribute to the limited number of NK or NKT cells found in NOD mice [3], something new and possibly interesting has been found among B lymphocytes. A sub-population of B cells, historically recognized only for their ability to generate antibodies, has been recently described as having regulatory properties (Bregs), similar yet different to the ones of Tregs with whom they seem to interact. Rituximab, a humanized monoclonal antibody against CD20, a marker of B cells, seems to give an advantage to Breg cell survival when administered to diabetic patients. The resulting Breg cell survival is attributed to the assumption that rituximab does not kill B lymphocyte precursors or plasma cells. This may explain the relative success obtained in a rituximab-based diabetes trial promoted by TrialNet [6]. As with using anti-CD3 antibodies, the partial or total, yet temporary, deletion of only one cell population was insufficient to achieve long lasting clinical results, substantiating the complexity of the orchestrated autoimmune attack generated against the pancreatic islets [7].
 |
 |
Figure 1. A citadel under siege: the peri-insulitis. Insulin (green), CD4+ and CD8+ T cells (red), cell nuclei (blue). |
|
In this issue of The Review of Diabetic Studies, Phillips et al. present a new immune intervention approach designed to target both CD4+ and CD8+ T cells by non-depleting antibodies. They found that this regimen possibly reverses the disease, not only in NOD mice, but also in humans [5]. These non-depleting antibodies seem to induce tolerance to their own Fc region, which preserves the possibility of repeated injections. Both anti-CD3 and non-depleting anti-CD4 have been shown to induce Tregs through a response dependent on TGFβ. Other examples of Treg-driven tolerance depends on IL-10 expression. Elevated IL-10 expression and Treg cell induction can be achieved by treatment with non-depleting anti-CD8 antibodies on the one hand, and TGF-β-dependent anti-CD3 and anti-CD4 antibody treatment on the other hand. The understanding of the possibly complementary mechanisms, by which these therapeutic approaches mediate diabetes reversal, should lead to the development of new inhibitors that could favor both inhibition of the autoreactive process and beta-cell recovery [5].
Dendritic cells
A new way to abrogate the autoimmune attack more efficiently is to exploit the mediating role of DC. These cells are capable of activating and maintaining immunoregulatory, "suppressive" cell networks. They are regulatory whilst remaining in a state of functional "immaturity". Functional immaturity can be conferred to DC partly by downregulating costimulatory pathways using systemic and molecule-specific approaches. Low-level expression of CD40, CD80, and CD86 can be induced by ex vivo treatment of DC from NOD mice with antisense oligodeoxynucleotides, AS-ODN, targeting the 5' ends of the respective primary transcripts. The administration of these ex vivo treated DC to syngeneic recipients can prevent the onset of disease and even revert already established diabetes. The safety of this approach is now being evaluated in a phase I clinical trial, in which autologous DC generated in vitro from leukapheresis products and treated with AS-ODN, are being re-administered to established type 1 diabetic adult patients [8]. When safety is proven, efficacy could be demonstrated by treating new onset patients to take advantage of both protection of remaining functional beta-cells and possibly the regenerative potential of a child's pancreas.
Beta-cell replacement and/or regeneration
The prevalent belief that the beta-cell mass is fixed by adulthood and that all adult beta-cells are fully differentiated is now being reexamined in light of recent studies. It has been shown that, albeit low, the endocrine pancreas has a regenerative capacity during disease progression. Some studies suggest that, although the physiological state of islet cells tends towards a fully differentiated phenotype, the lack of autoimmune aggression, together with "danger" signals generated by massive beta-cell destruction, may trigger processes inside progenitors. The processes, whether islet-resident or ductal epithelium-resident, result in some degree of insulin-producing cell regeneration [9]. The physiological equilibrium between lost and newly generated beta-cells can be altered by the action of those beta-cell-specific, autoreactive T cell clones. When the killing activity of diabetogenic T cells overcomes the combined effect of Tregs and the regenerative compensatory activity of the pancreatic gland, then the number of functional beta-cells progressively decreases. The decrease in beta-cell number then causes insulin secretion to fall below the critical level to maintain gluco-homeostasis in the body. After clinical onset of the disease, even if the regenerative properties of the pancreas remain functional, the continued presence of autoreactive T cells consistently nullifies the reparative effort. This could be the basis of the relapsing-remitting hypothesis originally proposed by von Herrath et al., which is discussed in their review published hereafter [2].
Islet allo-transplantation, a strategy to substitute the patients’ lost islets, has been proposed as a safe and efficient way to counteract autoimmunity and restore physiological insulin production. The prospect of better glucose control and fewer complications has considerable appeal compared to traditional glucose monitoring and daily insulin injection regimens. However, the long-term success of human islet transplantation has been limited, with ~90% of patients who achieved insulin independence requiring insulin again within 5 years post-transplant [10]. Even if a better immunosuppressive regimen can be found that will allow a longer graft survival, the limited number of pancreas donors together with the risk of allo-sensitization possibly jeopardizing future kidney transplantation in type 1 diabetic patients, will limit dramatically the clinical application of this procedure.
The need for data indicating the efficacy of alternative islet sources in non-human primate (NHP) models has been highlighted recently [11]. Successful xenotransplantation of porcine insulin-producing cells into diabetic patients could restore the physiological islet function, without the risk of allo-sensitization. A clue to this success is that pig insulin has been successfully used for years in treating diabetic patients. Long-term pig islet survival under a limited immunosuppressive protocol would significantly expand the clinical applicability of beta-cell replacement therapy for diabetes. The transplantation of islets from hCD46 pigs resulted in graft survival and insulin-independent normoglycemia in four of five monkeys for the 3 months follow-up of the experiment. One normalized recipient, selected at random, was followed for >12 months. Inhibition of complement activation by the expression of hCD46 on the pig islets did not substantially reduce the initial loss of islet mass; rather it was effective in limiting antibody-mediated rejection. This resulted in a reduced need for immunosuppression to preserve a sufficient islet mass to maintain normoglycemia long-term [12].
Until now, the liver has been the most commonly used site for islet transplantation, although survival of the graft after infusion into the portal vein (PV) has turned out to be suboptimal. Approximately one-third of islet recipients experience at least one adverse event within the first year, with almost half of these related to the transplant procedure itself. Also, exposure to blood results in a substantial loss of islets from the "instant blood-mediated inflammatory reaction" (IBMIR), a pathologic nonspecific inflammatory response to tissues that are not normally present in the blood. The gastric submucosal space (GSMS) seems to offer potential advantages over the PV as transplantation site. There is minimal risk of bleeding or thrombosis and, as the islets are not injected directly into the blood, IBMIR would be avoided. Importantly, the GSMS is endoscopically accessible, which would allow islet transplantations to be minimally invasive; also, repeated transplantation could readily be performed [13].
Another factor has become important following recent research results: the level of C-peptide secretion. C-peptide is the segment connecting insulin A and B chains. It is generated in pancreatic beta-cells as the natural product of pro-insulin cleavage. For a long time, it was considered biologically important only for favoring pro-insulin folding within the secretory granules of the beta-cells. However, recent studies have challenged this view by demonstrating that C-peptide is capable of biological effects in many different cell types. In endothelial and vascular smooth muscle cells exposed to hyperglycemia, C-peptide is internalized via early endosomes. Once inside the cell, it can reduce inflammatory damage mediated by nuclear factor (NF)-κB activation and upregulation of RAGE signaling [14, 15]. Beside relieving states of glucose instability and problems with hypoglycemia, islet transplantation promotes C-peptide secretion. It is maintained in the majority of patients for up to 5 years, even when most of them have reverted to using some insulin [10]. This limited yet steady supply of C-peptide seems sufficient to reduce a lot of the classical diabetes complications, re-proposing the possible biological activity of this peptide.
Breakthrough in diabetes therapy right around the corner?
In light of all of these successes a real cure for type 1 diabetes seems to be right around the corner. However, the corner seems to be ever elusive. Rather than becoming frustrated by the outstanding difficulties, we have to proceed with renewed determination towards our goal to definitively cure type 1 diabetes. We will model our resolve upon the waves of the sea, which ebb and flow and weaken and strengthen every day…
Conflict of interest statement: The authors declare that they have no competing conflict of interests.
References
- Tfayli H, Bacha F, Gungor N, Arslanian S. Phenotypic type 2 diabetes in obese youth: insulin sensitivity and secretion in islet cell antibody-negative versus -positive patients. Diabetes 2009. 58(3):738-744. [DOD] [CrossRef]
- Coppieters KT, von Herrath MG. Histopathology of type 1 diabetes: old paradigms and new insights. Rev Diabet Stud 2009. This issue. [DOD]
- Jordan MA, Poulton LD, Fletcher JM, Baxter AG. Allelic variation of Ets1 does not contribute to NK and NKT cell deficiencies in type 1 diabetes susceptible NOD mice. Rev Diabet Stud 2009. This issue. [DOD]
- Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J 2009. 28:2812-2824. [DOD]
- Phillips JM, Parish NM, Raine T, Bland C, Sawyer Y, De La Pena H, Cooke A. Type 1 diabetes development requires both CD4+ and CD8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev Diabet Stud 2009. This issue. [DOD]
- Peskovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker D, Gitelman S, Goland R, Gottlieb P, Marks JB, McGee P, Moran A, et al. Preservation of beta cell function by B-lymphocyte depletion with rituximab in patients with new onset autoimmune diabetes. N Engl J Med 2009. In press. [DOD]
- Herold KC, Gitelman S, Greenbaum C, Puck J, Hagopian W, Gottlieb P, Sayre P, Bianchine P, Wong E, Seyfert-Margolis V, et al. Treatment of patients with new onset type 1 diabetes with a single course of anti-CD3 mAb teplizumab preserves insulin production for up to 5 years. Clin Immunol 2009. 132(2):166-173. [DOD] [CrossRef]
- Phillips B, Giannoukakis N, Trucco M. Dendritic cell-based therapy in type 1 diabetes mellitus. Expert Rev Clin Immunol 2009. 5:325-339. [DOD] [CrossRef]
- Bottino R, Casu A, Criscimanna A, He J, Van Der Windt DJ, Rudert WA, Giordano C, Trucco M. Recovery of endogenous beta cell function in non-human primates following chemical diabetes induction and islet transplantation. Diabetes 2009. 58:442-447. [DOD] [CrossRef]
- Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, Lakey JR, Shapiro AM. Five-year follow-up after clinical islet transplantation. Diabetes 2005. 54:2060-2069. [DOD] [CrossRef]
- Ruggenenti P, Remuzzi A, Remuzzi G. Decision time for pancreatic islet-cell transplantation. Lancet 2008. 371:833-884. [DOD]
- van der Windt DJ, Bottino R, Casu A, Campanile N, Smetanka C, He J, Murase N, Hara H, Ball S, Loveland BE, Ayares D, Lakkis FG, Cooper DK, Trucco M. Long-term controlled normoglycemia in diabetic non-human primates after transplantation with hCD46 transgenic porcine islets. Am J Transplantation 2009. In press. [DOD]
- Echeverri GJ, McGrath K, Bottino R, Hara H, Dons EM, van der Windt DJ, Ekser B, Casu A, Houser S, Ezzelarab M, et al. Endoscopic gastric submucosal transplantation of islets (Endo-STI): technique and initial results in diabetic pigs. Am J Transplantation 2009. In press. [DOD]
- Sima AA, Zhang W, Kreipke CW, Rafols JA, Hoffman WH. Inflammation in diabetic encephalopathy is prevented by C-peptide. Rev Diabet Stud 2009. 6:37-42. [DOD] [CrossRef]
- Luppi P, Geng X, Cifarelli V, Drain P, Trucco M. C-peptide is internalized in human endothelial and vascular smooth muscle cells via early endosomes. Diabeteologia 2009. In press. [DOD]
|


)

