Original Data
| Rev Diabet Stud,
2010,
7(1):36-46 |
DOI 10.1900/RDS.2010.7.36 |
Glucose Homeostasis in Pre-diabetic NOD and Lymphocyte-Deficient NOD/SCID Mice During Gestation
Josiane Coulaud1, Sylvie Durant2, Francoise Homo-Delarche1
1Laboratoire B2PE (Biologie et Pathologie du Pancréas Endocrine), Unité de Biologie Fonctionnelle et Adaptative (BFA)-EAC CNRS 4413, Université Paris-Diderot, Paris, France
2Inserm UMR-S 747, Centre Universitaire, Université Paris-Descartes, France
Address correspondence to: Françoise Homo-Delarche, e-mail: francoise.homo-delarche@paris7.jussieu.fr
Manuscript submitted March 22, 2010; resubmitted April 19, 2010; accepted May 7, 2010.
Keywords: type 1 diabetes, NOD/SCID mouse, gestation, insulin, islet, glucagon, corticosterone, macrophage
Abstract
BACKGROUND: Unlike other strains, spontaneously type 1 non-obese diabetic (NOD) mice experience transient hyperinsulinemia after weaning. The same applies for NOD/SCID mice, which lack functional lymphocytes, and unlike NOD mice, do not develop insulitis and diabetes like NOD mice. AIMS: Given that β-cell stimulation is a natural feature of gestation, we hypothesized that glucose homeostasis is disturbed in gestate pre-diabetic NOD and non-diabetic NOD/SCID mice, which may accelerate the onset of diabetes and increase diabetes prevalence. METHODS: During gestation and postpartum, mice were analyzed under basal feed conditions, and following glucose injection (1 g/kg, i.p.) after overnight fast, using glucose tolerance test (GTT). Glycemia, corticosteronemia, blood and pancreatic insulin, glucagon levels, islet size, and islet morphology were evaluated. Glycemia and mortality were assessed after successive gestations in NOD mice mated for the first time at 2 different ages. RESULTS: 1. Basal glucagonemia rose markedly in first-gestation fed NOD mice. 2. β-cell hyperactivity was present earlier in first-gestation non-diabetic fasted NOD and NOD/SCID mice than in age-matched C57BL/6 mice, assessed by increased insulin/glucose ratio after GTT. 3. Overnight fasting increased corticosteronemia rapidly and sharply in pre-diabetic gestate NOD and NOD/SCID mice. 4. Islet size increased in non-diabetic gestate NOD mice compared with C57BL/6 mice. 5. Successive gestations accelerated diabetes onset, and contributed to increased mortality in NOD mice. CONCLUSIONS: First-gestation pre-diabetic NOD and non-diabetic NOD/SCID mice exhibited β-cell hyperactivity and deregulation of glucagon and/or corticosterone secretion. This amplified normally occurring insulin resistance, further exhausted maternal β-cells, and accelerated diabetes in NOD mice.
Abbreviations: 1 N HCL - 88,8 ml HCL are mixed with so much water that the dilution yields 1 l; ANOVA - analysis of variance; BM8+ - scavenger macrophage marker; C57BL/6 - inbred strain C57 black 6; CD4 - cluster of differentiation 4 (glycoprotein expressed on Th, Treg, and dendritic cells; co-receptor assisting the T cell receptor); D1PP - day 1 postpartum; D7PP - day 7 postpartum; FoxP3 - forkhead box P3 (transcriptional activator); g - g force, also called "relative centrifugal field" (RCF); conversion to rounds per minute (rpm): RCF = 11.18 (rcm) (rpm/1000)2, with rcm = radius of the rotor in centimeters; GAD - glutamic acid decarboxylase; G7 - day 7 of gestation; G14 - day 14 of gestation; G18 - day 18 of gestation; GTT - glucose tolerance test; HCL - hydrochloric acid; HPA axis - hypothalamo-pituitary-adrenal axis; IL-1 - interleukin-1; i.p. - intraperitoneal; NaCl - sodium chloride; NOD - nonobese diabetic; RIA - radioimmunoassay; SCID - severe combined immunodeficiency (mouse model without functional T and B cells); SEM - standard error of mean; T1D - type 1 diabetes; T2D - type 2 diabetes; % vol/vol - percent volume per volume
Introduction
The non-obese diabetic (NOD) mouse model is a well-recognized model of type 1 diabetes (T1D). This model provides the basis for extensive genetic and immunological investigations. It is commonly assumed that progressive infiltration of the islets of Langerhans by macrophages and dendritic cells starts at weaning, rapidly followed by T cell migration around and into the islets (peri-insulitis and insulitis, respectively). This process leads to progressive β-cell destruction and finally diabetes. However, little is known about neuroendocrine aspects of the disease [1]. Previously, we reported that, unlike other strains, pre-diabetic 8-week-old NOD mice experience transient basal hyperinsulinemia that starts at weaning, concomitantly with marked macrophage and dendritic cell infiltration [1-4]. The same applies for NOD/SCID mice, which lack functional lymphocytes and do not develop insulitis and diabetes. In 8-week-old NOD mice, this β-cell hyperactivity coexists with mild insulin resistance [2].
Interestingly, islets of neonates born from NOD dams, exhibit various alterations of islet immune and endocrine cells, whilst controls do not show these characteristics. Among these alterations are signs of β-cell hyperactivity, α-cell anomalies, increased numbers of antigen-presenting cells, and/or disturbances of extracellular matrix proteins [1, 5-8]. Some of these alterations are also observed in NOD/SCID mice [5, 6, 8]. Gestation is known to rapidly induce a maternal physiological state of β-cell stimulation to promote fetal growth, and later some degree of maternal insulin resistance [9]. In addition, the in utero programming of metabolic disturbances (e.g., insulin resistance, obesity, hypercholesterolemia, and T2D) and cardiovascular disease is more and more documented in animals and humans [9-11].
Therefore, we hypothesized that the endocrine environment might be altered during gestation in pre-diabetic NOD mice and lymphocyte-deficient non-diabetic NOD/SCID mice. To verify this hypothesis, we examined the effect of gestation on maternal glucose homeostasis and islet parameters in these 2 strains of mice compared with C57BL/6 control mice.
Materials and methods
Animals and experimental procedures
NOD, NOD/SCID, and C57BL/6 mice were bred under specific pathogen-free conditions at the facilities of Necker Hospital, Paris, France. Animal facilities and care were in accordance with accepted standards, established by the French National Centre for Scientific Research. NOD, NOD/SCID, and C57BL/6 females were mated; the morning appearance of the vaginal plug was taken as day 0 of gestation. At least twice as many females as needed for a given experiment were mated for the first time at 9 weeks of age, except where otherwise stated. The animals were studied at different days of gestation (G7, G14, and G18), and on day 1 and/or 7 postpartum (D1PP, D7PP). For comparison of the 3 strains, mice were mated during a narrow time period to minimize possible environmental disturbances on long-term investigation [1].
The study design included the following examinations: 1. The alteration of various parameters of glucose homeostasis (glycemia, insulinemia, glucagonemia, corticosteronemia, and pancreatic contents of insulin and glucagon) under basal feed conditions in first-gestation NOD and C57BL/6 (control) mice at G7, G14, G18, and day 7 postpartum (D7PP). 2. The alteration of glycemia, insulinemia, and corticosteronemia during glucose tolerance test (GTT), i.e. after intra-peritoneal (i.p.) glucose injection (1 g/kg of body weight, dissolved in 0.9% NaCl, Assistance Publique, France [2]) or NaCl (vehicle-injection) in first-gestation fasted NOD, NOD/SCID, and C57BL/6 mice. For comparison, fasted never-gestate 12-week-old mice of the 3 strains were also evaluated by GTT. 3. Islet size and islet morphology at D1PP in NOD versus C57BL/6 mice was studied, because gestation modifies islet characteristics [12, 13]. 4. The effect of successive gestations on diabetes onset and survival in two groups of NOD mice was verified after first mating at 6 or 10 weeks of age, and subsequently after matings at 3-4 week intervals.
All experiments started at 9:00 am on mice that had not been subjected to previous handling. Non-anesthetized animals were bled in less than 2 min by retro-orbital puncture. As previously published, this technique avoids stress-induced metabolic changes [14]. As in other endocrine studies in NOD mice, different groups of animals were used per condition examined (strain, time, injection) to avoid stress effects on glucose homeostasis [2, 5, 14-16]). At least six or more mice per condition were planned. In some cases, it could be less (4-5/group), because of the variable recovery rate of gestate mothers. To assure comparability of GTT responses of the 3 strains, we took the measurement 5 min after glucose injection, according to previous data [2].
Blood samples, supplemented with recombinant aprotinin (30,000 kIU/ml, Bayer Inc., USA) were kept on ice, then centrifuged at 13,000 × g for 2 min at 4°C, and stored at -20°C. After bleeding, mice were immediately killed by cervical dislocation. Pancreases were rapidly removed, weighed, and homogenized in 15 ml of cold acidified ethanol extraction medium (1.5% (vol/vol) 1 N HCl in 75% ethanol). After addition of another 10 ml of extraction medium, the homogenates were centrifuged (800 × g for 15 min, 4°C), and the supernatants were left standing overnight at 4°C. The pH of the supernatants was adjusted to 8.5 with ammonium hydroxide, and after centrifugation (800 × g for 15 min, 4°C), 5 ml of each supernatant were stored at -20°C until assayed.
Glucose, insulin, and glucagon measurements
Glycemia was measured using the glucose-oxidase method (Biotrol glucose enzymatic color, Biotrol, France). Insulin, glucagon, and corticosterone concentrations were determined by standard radioimmunoassay (RIAs: SB-insulin-CT, CIS Biointernational, Biodata, Pharmacia, and ICN Biomedicals Inc., Sorin Biomedica, France), as described elsewhere [2, 5].
Islet measurement and immunohistochemistry
For macrophage marker immunohistochemistry, pancreases were embedded in Tissue-Tek (Miles, USA), and frozen in dry-ice chilled isopentane at day 14 of maternal gestation. Cryostat sections were fixed for 10 min in acetone containing 0.03% hydroperoxide and labeling was performed, as previously described [6]. For hematoxylin-eosin staining, pancreases were excised from NOD mothers 1 day after delivery, and fixed in Bouin's fluid, followed by buffered 10% formalin, and processed for paraffin embedding. Islet surfaces were then assessed via a VIDAS-RT image analysis system (Kontron Elektronik GmbH/Carl Zeiss, The Netherlands). The total number of pancreas sections was counted for a given strain (n = 6 mice/strain). Only one section per mouse pancreas was measured to ensure that an individual islet was not measured twice [5, 6, 17]. The total number of islets counted was around 150-160 per strain. For islet surface measurement, only the endocrine cell contribution was measured (excluding the immune cell contribution (insulitis) to the islet area). Data were expressed in pixels. The conversion factor from pixel to mm2 was 0.0001113.
Statistical analysis
All data are means ± SEM. In most experiments, possible effects of strain, age, and/or injection were analyzed using two- or three-way p-factor analysis of variance (ANOVA). As usually published [2, 5, 15, 16], post-hoc analysis was performed, using the Newman-Keuls test, when effects and interactions were significant (p < 0.05), as assessed by ANOVA. Student t-test, Kaplan-Meier, and Log-Rank tests were used in punctual experiments. Significance was defined as p < 0.05, and trend towards significance 0.05 < p < 0.1.
Results
Parameters of glucose homeostasis during first gestation and first week postpartum in fed non-diabetic NOD mice vs. C57BL/6 controls
Figure 1A shows that NOD mice became progressively heavier than age-matched controls during gestation. They were significantly heavier before and after birth (G18 and D7PP, p < 0.001 in both cases). In fed pre-diabetic NOD mice (Figure 1B), basal glycemia declined significantly from G7 to G18 (p = 0.006), but rose from G18 to D7PP (p = 0.01). At G18, glycemia was significantly lower in NOD mice than in C57BL/6 controls (p = 0.004). NOD pancreatic insulin contents (Figure 1E), but not basal insulinemia (Figure 1C), varied significantly throughout gestation and lactation. It increased from G7 to G18 (p = 0.015), and showed decreasing trend afterwards (p = 0.05). NOD basal glucagonemia (Figure 1D) increased progressively and significantly during gestation and postpartum (p-values varied between each time-point from <0.005 to 0.05). NOD glucagonemia was significantly higher than in controls at G14, G18, and D7PP (p varied from <0.0001 to 0.02). In contrast, NOD pancreatic glucagon content (Figure 1F) was significantly lower than that of C57BL/6 mice at G7 (p = 0.01), and showed a decreasing trend at G14 and D7PP (p = 0.06 in both cases).
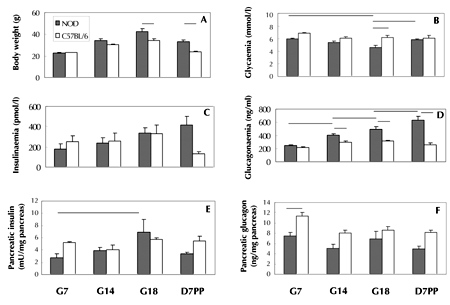 |
 |
Figure 1. Body weight and basal parameters of glucose homeostasis during the first gestation and first week postpartum in non-diabetic fed NOD mice vs. C57BL/6 controls. Mice were studied on G7, G14, G18, and day 7 postpartum (D7PP). Panel A: Body weight in NOD vs. C57BL/6 mice on G18 and D7PP, p < 0.001 in both cases. NOD glycemia (panel B): decreased from G7 to G18, p = 0.006; increased from G18 to D7PP, p = 0.01; at G18, lower glycemia in NOD mice than in controls, p = 0.004. NOD glucagonemia (panel D): increased from G7 to G14, G14 to G18, and G18 to D7PP, p-values varying from < 0.005 to < 0.05. Pancreatic NOD insulin (panel E): increased from G7 to G18, p = 0.015. Decreased pancreatic NOD vs. C57BL/6 glucagon contents at G7, p = 0.01 (panel F). Data are mean ± SEM, n = 4-5 mice per strain and gestation/lactation day. Lines indicate the main statistically significant differences. |
|
Glucose tolerance test in first-gestation fasted non-diabetic NOD, lymphocyte-deficient NOD/SCID, and control C57BL/6 mice
Figure 2 (panel A) shows the glycemic response 5 min after glucose injection (1 g/kg of body weight i.p.). On G7, G14, and G18, the glycemic indexes (expressed as glucose to NaCl injection ratios) were 2.5, 2.6, and 2.4 for NOD, 2.4, 2.3, and 1.7 for NOD/SCID, and 2.0, 1.9, and 1.2 for C57BL/6 mice, respectively. These data showed that NOD and NOD/SCID mice had higher glycemic responses during GTT than C57BL/6 controls. Insulin responses to NaCl injection alone were similar among the 3 strains during gestation (Figure 2, panel B), but insulin responses to glucose administration differed. Indeed, blood insulin/glucose ratios (Figure 2, panel C) were significantly higher in NOD/SCID vs. NOD on G7 (p = 0.01), in NOD/SCID and NOD vs. C5BL/6 on G7 and G14 (p = 0.0004 and p = 0.01, respectively), and in C57BL/6 mice on G18 vs. G7 (p = 0.04). These differences suggested an early versus late gestation-induced insulin resistance in the three strains (NOD, NOD/SCID, and C57BL/6), i.e. NOD and NOD/SCID mice had an early and C57BL/6 mice a late insulin resistance.
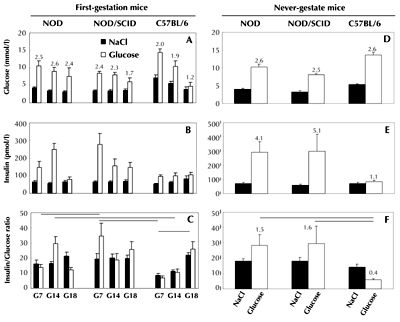 |
|
Figure 2. Glucose tolerance test in first-gestation and never-gestate non-diabetic NOD, lymphocyte-deficient NOD/SCID, and control C57BL/6 mice. Glucose (1 g/kg body weight, i.p.) was injected into overnight fasted NOD, NOD/SCID, and C57BL/6 mice, which had been mated at 9 weeks of age (panels A-C), or 12-week-old never-gestate mice of the 3 strains (panels D-F). Blood parameters were measured 5 min after glucose injection. Glucose-induced insulinemia/glycemia ratios in first-gestation mice (panel C): on G7, NOD vs. NOD/SCID, p = 0.01, and NOD/SCID vs. C57BL/6, p = 0.0004; on G14, NOD vs. C57BL/6, p = 0.01; C57BL/6 mice on G18 vs. G7, p = 0.04 (mean ± SEM, n = 5-7 mice per strain and gestation day). Glucose-induced insulinemia/glycemia ratios in never-gestate 12-week-old mice (panel F): NOD and NOD/SCID mice vs. C57BL/6 controls, p = 0.03 in both cases. Data are mean ± SEM. NOD, NOD/SCID, and C57BL/6 mice (n = 7-9) and (n = 5) per strain/gestation day/type of injection, respectively. Lines indicate the main statistically significant differences. |
|
Glucose tolerance test in never-gestate 12-week-old fasted non-diabetic NOD, NOD/ SCID, and control C57BL/6 mice
As shown in Figure 2 (panel D), glucose/NaCl indexes 5 min after NaCl injection were similar in all three strains (NOD = 2.6, NOD/SCID = 2.5, and C57BL/6 = 2.6), suggesting comparable glycemic responses. Insulin responses to NaCl injections were also similar among the 3 strains, but glucose-induced insulinemia/NaCl indexes (Figure 2, panel E) were markedly higher in NOD and NOD/SCID mice than in C57BL/6 controls (4.1, 5.1, and 1.1, respectively). Therefore, insulinemia/glycemia ratios (Figure 2, panel F) were significantly higher in non-diabetic NOD and NOD/SCID than in C57BL/6 mice (1.5, 1.6 versus 0.4, respectively; p = 0.03 in both cases). These data showed that never-gestate, 12-week-old mice with the NOD genetic background were insulin resistant.
Circulating corticosterone levels in first-gestation non-diabetic NOD, NOD/SCID, and control C57BL/6 mice subjected to the various experimental conditions
Fasting corticosteronemia of never-gestate 12-week-old females of the 3 strains were similar (300-400 ng/ml; data not shown). Figure 3 (panel A) shows as expected that corticosteronemia increased progressively under basal feed conditions, without injection during gestation [18]. In all three strains, it increased in similar courses to 400-600 ng/ml (G7 vs. G14, p < 0.05, and G14 vs. G18, p < 0.001 for all strains).
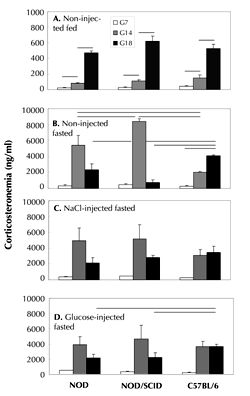 |
|
Figure 3. Circulating corticosterone in first-gestation non-diabetic NOD, NOD/SCID, and control C57BL/6 mice subjected to various experimental diet and injection conditions. Corticosteronemia was measured on G7, G14, and G18. Non-injected fed mice (panel A): G7 vs. G14, p < 0.05, and G14 vs. G18, p < 0.001 for all strains, respectively (mean ± SEM, 4 mice/strain/gestation day). Non-injected fasted mice (panel B): at G14, NOD and NOD/SCID mice vs. C57BL/6 controls, p = 0.05 and p = 0.0005, respectively, and NOD/SCID vs. NOD mice, p = 0.01. Fasted and i.p. NaCl- or glucose-injected conditions (panels C and D, respectively): at G18, glucose-injected NOD and NOD/SCID mice vs. C57BL/6, p < 0.05 in both cases. In panels B-D: data are mean ± SEM, n = 10-15 mice/strain and gestation day. Lines indicate the main statistically significant differences. |
|
During gestation and after overnight fasting without injection (Figure 3, panel B), C57BL/6 corticosterone levels rose progressively to 4000 ng/ml from G7 to G18 (G7 vs. G18, p = 0.005). In NOD and NOD/SCID mice, corticosteronemia peaked earlier (at G14) and at significantly higher levels than in controls (6000 and 8000 ng/ml, p = 0.05 and 0.005, respectively). At G18, corticosterone levels were markedly lower in NOD and NOD/SCID mice than in controls (p = 0.05 and p = 0.01, respectively). Moreover, corticosteronemia at G14 was significantly higher in non-injected fasted NOD/SCID than NOD mice (p = 0.01).
Figure 3, panels C and D, show blood corticosterone profiles in fasted mice of the 3 strains after NaCl and glucose injection, respectively. NOD and NOD/SCID corticosteronemia peaked at G14, as in their non-injected fasted counterparts (Figure 3, panels C and D vs. B). However, G14 corticosterone levels were lower for injected (around 5000 ng/ml) than for non-injected NOD/SCID mice (8000 ng/ml). At G18 (Figure 3, panel D), corticosteronemia in NOD and NOD/SCID mice was significantly lower than in C57BL/6 mice (p < 0.05 in both cases).
Characteristics of maternal islets during gestation and at delivery
Figure 4 shows immunohistochemical labeling of BM8+ and ER-MP23+ macrophage infiltration in C57BL/6 and NOD pancreases at G14. Similar as previously published in never-gestate mice [6], during gestation macrophage infiltration was more distinct around NOD islets than around those of controls.
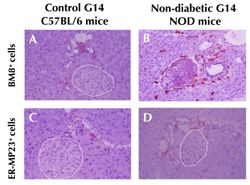 |
|
Figure 4. Macrophage labeling on pancreas sections from first-gestation G14 C57BL/6 controls and non-diabetic NOD mice. Presence of few BM8+ and ER-MP23+ macrophages around islets (white dashed lines) and ducts of C57BL/6 controls (A and C). More macrophages were observable in non-diabetic NOD pancreases during gestation (B and C). Magnification ×280. |
|
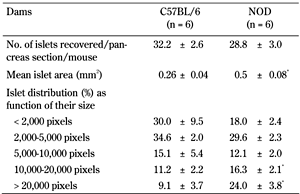
Mean islet size and islet distribution, as function of islet size, was assessed in first-gestation C57BL/6 and NOD pancreases at D1PP (Table 1). NOD dams had significantly larger mean islet areas and higher mean percentages of the largest islets (10,000-20,000 and >20,000 pixels) than C57BL/6 dams (p < 0.05 in all cases).
Table
1.
Characteristics of control C57BL/6 and non-diabetic NOD dam islets 1 day after delivery |
|
 |
|
Legend:
Data are mean ± SEM, Student t-test. *p < 0.05. |
|
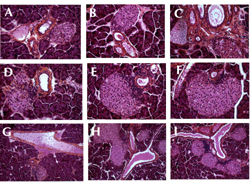
Figure 5 illustrates variable-sized islets associated with ducts, which is characteristic of islets in neogenesis. Insulitis could be observed at the ductal poles of islets (Figure 5B-F and I). In some cases, ducts were associated with several infiltrated islets (Figure 5H and 5I). Sometimes, numerous mononuclear cells were observed in vessels near duct/islet structures (Figure 5G).
 |
|
Figure 5. Hematoxylin-eosin staining of non-diabetic NOD dam pancreas sections one day after delivery. Non-diabetic NOD dam islets of various sizes at birth. Some islets appeared to be free of insulitis (A), others showed insulitis at the duct-insular structure (C-F, and I). Magnification A-F: ×280; G-I: ×140. Other islets were almost completely invaded by insulitis (data not shown). |
|
Effect of successive gestations on diabetes
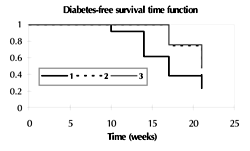
Two groups of pre-diabetic NOD females were mated at every time starting at 6 weeks (group 1 with 5 successive gestations, n = 13) or 10 weeks (group 2 with 4 successive gestations, n = 8), and compared to never-gestate mice from the same colony (group 3, n = 46) within the same period of time (Figure 6). The mean survival time from birth onwards and without diabetes was 17.1 ± 1.0, 20.0 ± 0.6, and 20.0 ± 0.2 weeks, for groups 1 to 3, respectively (Kaplan-Meier analysis). A significant diabetes-free difference in the survival rate was observed among the 3 groups (p = 0.02) (Figure 6). Finally, there was a marked trend towards survival difference between group 1 and 2 (p = 0.06, Log-Rank test). Therefore, it seems that repeated mating and/or age at first mating influence diabetes onset and survival.
 |
|
Figure 6. Effect of successive gestations on diabetes onset and survival in NOD mice mated at 2 different ages. Two groups of pre-diabetic NOD females were mated at every time starting at 6 weeks (group 1, 5 successive gestations, n = 13) or 10 weeks (group 2, 4 successive gestations, n = 8), and compared to never-gestate mice from the same colony within the same period of time (group 3, n = 46). The diagram shows a significant diabetes-free survival difference among the 3 groups (p = 0.02, Kaplan-Meier analysis). |
|
Discussion
Pregnancy is known to modulate glucose homeostasis and to induce gestational diabetes, possibly followed by permanent maternal diabetes [19-22]. Also, pregnancy in diabetic mothers may be associated with intrauterine death, perinatal mortality, and birth weight anomalies [23, 24]. Pregnancy is a natural state of β-cell hyperactivity and insulin resistance, which may decompensate according to maternal genetic background and/or environment [19].
β-cell hyperactivity and insulin resistance are two well-recognized components of T2D pathophysiology. However, β-cell hyperactivity may also play a role in T1D pathogenesis [1, 3, 4]. Hyperactive endocrine cells are more prone to autoimmune reactions, because of higher levels of autoantigens, adhesion and MHC molecules, and higher sensitivity to cytokine-induced damage [4, 25-27]. The existence of transient β-cell hyperactivity, triggered by a state of insulin resistance, is able to accelerate disease progression, which was documented in humans with T1D [28-33]. Previously, we have described a state of β-cell hyperactivity beginning at weaning in the spontaneous NOD mouse model, when diet shifts from maternal milk (rich in fatty acids and low in carbohydrates) to laboratory chow (poor in fatty acids and rich in carbohydrates) [1-3]. This β-cell hyperactivity coexists with mild insulin resistance in 8-week-old NOD mice [2]. Comparable with a state of sustained and/or repetitive β-cell stimulation, gestations seem to accelerate the onset of diabetes and increased its prevalence in non-diabetic NOD mice.
To confirm the presence of this maternal β-cell stimulation, we conducted glucose tolerance tests (injection of 1 g glucose per kg body weight, i.p.). It showed that β-cell hyperactivity was present under basal feed conditions in first-gestation age-matched (12-week-old) non-diabetic NOD compared to C5BL/6 controls. This conclusion is based on the following observations:
1. Decreased glycemia during gestation after an initial increase at G7.
2. Trend towards increased insulinemia associated with progressively increased pancreatic insulin content. Notably, first-gestation NOD mice exhibited higher percentages of large islets than first-gestation controls, and showed signs of islet neogenesis, which is in fact not impaired in the NOD mouse [34, 35].
Basal glucagonemia rose progressively in first-gestation non-diabetic fed NOD, but not in C57BL/6 mice. Whilst NOD pancreatic glucagon content was lower than in controls. Glucagon anomalies before and after birth were observed independently of the age of NOD mothers at mating (unpublished data).
Glucagon, an adaptive hormone to counterbalance hypoglycemia, increases during pregnancy in normal and gestational diabetic women [36]. Anomalies of circulating glucose-induced glucagonemia, and a physiological α-cell insulin resistance, have been noted in gestational (or post-gestational) diabetic mothers compared to normal mothers during pregnancy and the postpartum period [37]. In non-diabetic gestate NOD mice, there are several reasons why hyperglucagonemia can result from an immune/endocrine imbalance in islets or the periphery. First, as insulin affects glucagon secretion [38, 39], α-cell disturbance may be primary or secondary to β-cell anomalies or insulin signaling. Conversely, as glucagon stimulates insulin secretion [40-42], the relative hyperglucagonemia may be partially responsible for the basal hyperinsulinemia. Second, cytokines such as interleukin-1 (IL-1), produced by the primary infiltrating immune cells, are known to stimulate glucagon secretion in vivo and in vitro [43]. Recently, IL-6 was shown to regulate α-cell mass expansion [44]. Notably, during normal pregnancy, systemic inflammatory responses induce cytokines [45]. This phenomenon could be aggravated in NOD mice, both in the periphery and within organs. In particular, inflammation is a feature in pancreatic islets of NOD mice, indicated by the presence of high amounts of immune cells. We observed this phenomenon in the present study. Third, NOD hyperglucagonemia may partly reflect the progressive loss of normal inhibition of glucagon secretion by the peri-islet GABAergic (glutamic acid decarboxylase (GAD)-containing) innervation. This disappears with age in NOD mice, in parallel with the progression of insulitis [46, 47]. Similarly, others have described early destruction of peri-islet Schwann cells in NOD mice, but not in NOD/SCID [48].
Pregnancy affects the hypothalamo-pituitary-adrenal (HPA) axis leading to increased glucocorticoids and ACTH levels during gestation [49]. The HPA response to fasting was abnormally stimulated in gestating NOD and NOD/SCID mice, and peaked around G14 instead of the end of gestation, as usually described in controls [18]. Moreover, non-injected fasting NOD, and particularly NOD/SCID mice, showed exaggerated responses at G14 compared to controls. However, at G18, the corticosterone response in non-injected fasting NOD mice, and particularly NOD/SCID mice, appeared to be lower than in controls, being the same as after glucose injection. These observations suggest abnormal regulation of the HPA axis in mice with the NOD genetic background, possibly involving a rapid down-regulation of corticosterone secretion in response to supplementary stress (e.g., i.p. injection after overnight fasting). In this context, NOD mice had fewer islet glucocorticoid receptors at 3 weeks of age [50]. When circulating glucose, corticosterone, and IL-6 responses to restraint were compared between various control strains (C57BL/6, DBA/2, and BALB/c) and pre-diabetic NOD mice at 8 weeks of age, the two strains with the highest corticosterone responses (BALB/c and NOD) had the lowest hyperglycemic and the highest IL-6 responses [16]. Due to the well-known hyperglycemic and anti-inflammatory/ immunosuppressive effects of glucocorticoids, these data suggest a certain degree of cortico-resistance in the two strains. Also, other HPA axis-related anomalies exist in NOD neonates before weaning, e.g. absence of stress hyporesponsive period, particularly after IL-1 injection [1], and early depressed dentate gyrus cell proliferation [51]. Finally, during gestation, HPA axis anomalies are more distinct after fasting in NOD/SCID than NOD mice. In this regard, athymic nude mice exhibit HPA axis alterations which are normalized by immune reconstitution with CD4+ lymphocytes [52].
Interestingly, some of the NOD gestational endocrine features are shared by NOD/SCID mice: high glycemic response, high insulinemia and signs of insulin resistance (increased insulin/glucose ratio) in response to glucose injection. Indeed, the NOD strain shares a common origin with the NON strain, a pre-T2D model that offers the possibility of exploring some common features such as insulin resistance [53]. Insulin resistance appears to peak earlier in NOD/SCID than NOD mice (G7 vs. G14). Various glucose-homeostasis anomalies have been described in lymphocyte-deficient mice: 1. Athymic nude mice exhibit several endocrine anomalies, including impaired glucose tolerance at very young age and peripheral insensitivity to insulin [54]. 2. Defects in regulatory CD4 Foxp3 cells appear to be linked to insulin resistance [55, 56]. 3. Gene studies of autoimmune-prone NOD/SCID and non-autoimmune-prone C57BL/6 mice suggest that, in the absence of hyperglycemia and β-cell destruction, the NOD genetic background may predispose to diabetic complications, including insulin resistance [57].
In conclusion, these data demonstrate endocrine disturbances during gestation even in pre-diabetic NOD mice. Maternal endocrine anomalies may be responsible for fetal and neonatal endocrine disturbances for the following reasons: 1. NOD mice produced by embryo transfer into ICR or DBA/2 mothers had a very low incidence of T1D, with only about 7% of the progeny developing diabetes by 40 weeks of age [58]. 2. NOD fetal pancreatic organ cultures produce more insulin that those from C57BL/6 or BALB/c controls [59]. 3. At birth, both sexes of NOD neonates exhibit signs of β-cell hyperactivity and α-cell anomalies, and maternal insulin treatment lowers β-cell hyperactivity in female neonates [5, 8]. 4. These neonatal islet anomalies are associated with increased numbers of some types of antigen-presenting cells and/or disturbance of extracellular matrix proteins in NOD and/or NOD/SCID pancreases [6, 7]. 5. Neonatal injection of glucose and arginine leads to diabetes enhancement and increased islet autoantigen expression (ICA and GAD 67) only in NOD females [60]. 6. Prenatal stress during the last week of gestation accelerates diabetes onset and increases in its prevalence in the NOD female progeny [61], while separation of NOD neonates from their mothers increases the risk of developing diabetes in both sexes [62]. It was suggested in the latter study that overstimulation of the HPA axis and induction of insulin resistance led to overloaded β-cells. Others have demonstrated that NOD mice have different risks for developing T1D depending on the strain of their foster mothers [63].
In summary, these data suggest that diabetes incidence in the NOD mouse is influenced by prenatal and postnatal maternal endocrine environment and/or behavior.
Acknowledgments:
This research was supported by grants from CNRS, Université Paris V, INSERM-NWO, BIOMED "Betimmune", and 5th PCRD "Monodiab".
References
- Homo-Delarche F. Neuroendocrine immuno-ontogeny of the pathogenesis of autoimmune disease in the nonobese diabetic (NOD) mouse. Ilar J 2004. 45:237-258. [DOD]
- Amrani A, Durant S, Throsby M, Coulaud J, Dardenne M, Homo-Delarche F. Glucose homeostasis in the nonobese diabetic mouse at the prediabetic stage. Endocrinology 1998. 139(3):1115-1124. [DOD] [CrossRef]
- Rosmalen JG, Leenen PJ, Pelegri C, Drexhage HA, Homo-Delarche F. Islet abnormalities in the pathogenesis of autoimmune diabetes. Trends Endocrinol Metab 2002. 13:209-214. [DOD] [CrossRef]
- Homo-Delarche F, Boitard C. Autoimmune diabetes: the role of the islets of Langerhans. Immunol Today 1996. 17:456-460. [DOD] [CrossRef]
- Pelegri C, Rosmalen JG, Durant S, Throsby M, Alves V, Coulaud J, Esling A, Pleau JM, Drexhage HA, Homo-Delarche F. Islet endocrine-cell behavior from birth onward in mice with the nonobese diabetic genetic background. Mol Med 2001. 7:311-319. [DOD]
- Charre S, Rosmalen JG, Pelegri C, Alves V, Leenen PJ, Drexhage HA, Homo-Delarche F. Abnormalities in dendritic cell and macrophage accumulation in the pancreas of nonobese diabetic (NOD) mice during the early neonatal period. Histol Histopathol 2002. 17:393-401. [DOD]
- Geutskens SB, Homo-Delarche F, Pleau JM, Durant S, Drexhage HA, Savino W. Extracellular matrix distribution and islet morphology in the early postnatal pancreas: anomalies in the non-obese diabetic mouse. Cell Tissue Res 2004. 318:579-589. [DOD] [CrossRef]
- Throsby M, Coulaud J, Durant S, Homo-Delarche F. Increased transcriptional preproinsulin II beta-cell activity in neonatal nonobese diabetic mice: in situ hybridization analysis. Rev Diabet Stud 2005. 2:75-83. [DOD] [CrossRef]
- Melnik BC. Permanent impairment of insulin resistance from pregnancy to adulthood: the primary basic risk factor of chronic Western diseases. Med Hypotheses 2009. 73:670-681. [DOD] [CrossRef]
- Le Clair C, Abbi T, Sandhu H, Tappia PS. Impact of maternal undernutrition on diabetes and cardiovascular disease risk in adult offspring. Can J Physiol Pharmacol 2009. 87:161-179. [DOD] [CrossRef]
- Palinski W, Nicolaides E, Liguori A, Napoli C. Influence of Maternal Dysmetabolic Conditions During Pregnancy on Cardiovascular Disease. J Cardiovasc Transl Res 2009. 2:277-285. [DOD] [CrossRef]
- Aerts L, Vercruysse L, Van Assche FA. The endocrine pancreas in virgin and pregnant offspring of diabetic pregnant rats. Diabetes Res Clin Pract 1997. 38:9-19. [DOD] [CrossRef]
- Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010. In press. [DOD]
- Amrani A, Chaouloff F, Mormede P, Dardenne M, Homo-Delarche F. Glucose, insulin, and open field responses to immobilization in nonobese diabetic (NOD) mice. Physiol Behav 1994. 56:241-246. [DOD] [CrossRef]
- Durant S, Geutskens S, Van Blokland SC, Coulaud J, Alves V, Pleau JM, Versnel M, Drexhage HA, Homo-Delarche F. Proapoptosis and antiapoptosis-related molecules during postnatal pancreas development in control and nonobese diabetic mice: relationship with innervation. Lab Invest 2003. 83:227-239. [DOD]
- Harizi H, Homo-Delarche F, Amrani A, Coulaud J, Mormede P. Marked genetic differences in the regulation of blood glucose under immune and restraint stress in mice reveals a wide range of corticosensitivity. J Neuroimmunol 2007. 189:59-68. [DOD] [CrossRef]
- Rosmalen JG, Pigmans MJ, Kersseboom R, Drexhage HA, Leenen PJ, Homo-Delarche F. Sex steroids influence pancreatic islet hypertrophy and subsequent autoimmune infiltration in nonobese diabetic (NOD) and NODscid mice. Lab Invest 2001. 81:231-239. [DOD]
- Mastorakos G, Ilias I. Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann N Y Acad Sci 2003. 997:136-149. [DOD] [CrossRef]
- Leturque A, Burnol AF, Ferre P, Girard J. Pregnancy-induced insulin resistance in the rat: assessment by glucose clamp technique. Am J Physiol 1984. 246:E25-E31. [DOD]
- Nieuwenhuizen AG, Schuiling GA, Moes H, Koiter TR. Role of increased insulin demand in the adaptation of the endocrine pancreas to pregnancy. Acta Physiol Scand 1997. 159:303-312. [DOD] [CrossRef]
- Lauenborg J, Hansen T, Jensen DM, Vestergaard H, Molsted-Pedersen L, Hornnes P, Locht H, Pedersen O, Damm P. Increasing incidence of diabetes after gestational diabetes: a long-term follow-up in a Danish population. Diabetes Care 2004. 27:1194-1199. [DOD] [CrossRef]
- Smirnakis KV, Martinez A, Blatman KH, Wolf M, Ecker JL, Thadhani R. Early pregnancy insulin resistance and subsequent gestational diabetes mellitus. Diabetes Care 2005. 28:1207-1208. [DOD] [CrossRef]
- Aerts L, Holemans K, Van Assche FA. Maternal diabetes during pregnancy: consequences for the offspring. Diabetes Metab Rev 1990. 6:147-167. [DOD] [CrossRef]
- Boloker J, Gertz SJ, Simmons RA. Gestational diabetes leads to the development of diabetes in adulthood in the rat. Diabetes 2002. 51:1499-1506. [DOD] [CrossRef]
- Aaen K, Rygaard J, Josefsen K, Petersen H, Brogren CH, Horn T, Buschard K. Dependence of antigen expression on functional state of beta-cells. Diabetes 1990. 39:697-701. [DOD] [CrossRef]
- Buschard K. The functional state of the beta cells in the pathogenesis of insulin-dependent diabetes mellitus. Autoimmunity 1991. 10:65-69. [DOD] [CrossRef]
- Bjork E, Kampe O, Karlsson FA, Pipeleers DG, Andersson A, Hellerstrom C, Eizirik DL. Glucose regulation of the autoantigen GAD65 in human pancreatic islets. J Clin Endocrinol Metab 1992. 75:1574-1576. [DOD] [CrossRef]
- Leslie RD, Taylor R, Pozzilli P. The role of insulin resistance in the natural history of type 1 diabetes. Diabet Med 1997. 14:327-331. [DOD] [CrossRef]
- Wilkin TJ. The accelerator hypothesis: weight gain as the missing link between Type I and Type II diabetes. Diabetologia 2001. 44:914-922. [DOD] [CrossRef]
- Greenbaum CJ. Insulin resistance in type 1 diabetes. Diabetes Metab Res Rev 2002. 18:192-200. [DOD] [CrossRef]
- Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia 2004. 47:1661-1667. [DOD] [CrossRef]
- Tsai EB, Sherry NA, Palmer JP, Herold KC. The rise and fall of insulin secretion in type 1 diabetes mellitus. Diabetologia 2006. 49:261-270. [DOD] [CrossRef]
- Xu P, Cuthbertson D, Greenbaum C, Palmer JP, Krischer JP. Role of insulin resistance in predicting progression to type 1 diabetes. Diabetes Care 2007. 30:2314-2320. [DOD] [CrossRef]
- O'Reilly LA, Gu D, Sarvetnick N, Edlund H, Phillips JM, Fulford T, Cooke A. Alpha-cell neogenesis in an animal model of IDDM. Diabetes 1997. 46:599-606. [DOD] [CrossRef]
- Suarez-Pinzon WL, Power RF, Yan Y, Wasserfall C, Atkinson M, Rabinovitch A. Combination therapy with glucagon-like peptide-1 and gastrin restores normoglycemia in diabetic NOD mice. Diabetes 2008. 57:3281-3288. [DOD] [CrossRef]
- Hornnes PJ. On the decrease of glucose tolerance in pregnancy. A review. Diabetes Metab 1985. 11:310-315. [DOD]
- Beis C, Grigorakis SI, Philippou G, Alevizaki M, Anastasiou E. Lack of suppression of plasma glucagon levels in late pregnancy persists postpartum only in women with previous gestational diabetes mellitus. Acta Diabetol 2005. 42:31-35. [DOD] [CrossRef]
- Dumonteil E, Magnan C, Ritz-Laser B, Meda P, Dussoix P, Gilbert M, Ktorza A, Philippe J. Insulin, but not glucose lowering corrects the hyperglucagonemia and increased proglucagon messenger ribonucleic acid levels observed in insulinopenic diabetes. Endocrinology 1998. 139:4540-4546. [DOD] [CrossRef]
- Kaneko K, Shirotani T, Araki E, Matsumoto K, Taguchi T, Motoshima H, Yoshizato K, Kishikawa H, Shichiri M. Insulin inhibits glucagon secretion by the activation of PI3-kinase in In-R1-G9 cells. Diabetes Res Clin Pract 1999. 44:83-92. [DOD] [CrossRef]
- Leiter EH, Coleman DL, Eppig JJ. Endocrine pancreatic cells of postnatal "diabetes" (db) mice in cell culture. In Vitro 1979. 15:507-521. [DOD] [CrossRef]
- Vecchio D, Luyckx A, Zahnd GR, Renold AE. Insulin release induced by glucagon in organ cultures of fetal rat pancreas. Metabolism 1966. 15:577-581. [DOD] [CrossRef]
- Kawai K, Yokota C, Ohashi S, Watanabe Y, Yamashita K. Evidence that glucagon stimulates insulin secretion through its own receptor in rats. Diabetologia 1995. 38:274-276. [DOD] [CrossRef]
- Zumsteg U, Reimers JI, Pociot F, Morch L, Helqvist S, Brendel M, Alejandro R, Mandrup-Poulsen T, Dinarello CA, Nerup J. Differential interleukin-1 receptor antagonism on pancreatic beta and alpha cells. Studies in rodent and human islets and in normal rats. Diabetologia 1993. 36:759-766. [DOD] [CrossRef]
- Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, Kerr-Conte J, Pattou F, Berney T, Pipeleers D, et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci U S A 2008. 105:13163-13168. [DOD] [CrossRef]
- Mendelson CR. Minireview: fetal-maternal hormonal signaling in pregnancy and labor. Mol Endocrinol 2009. 23:947-954. [DOD] [CrossRef]
- Saravia-Fernandez F, Faveeuw C, Blasquez-Bulant C, Tappaz M, Throsby M, Pelletier G, Vaudry H, Dardenne M, Homo-Delarche F. Localization of gamma-aminobutyric acid and glutamic acid decarboxylase in the pancreas of the nonobese diabetic mouse. Endocrinology 1996. 137:3497-3506. [DOD] [CrossRef]
- Saravia F, Homo-Delarche F. Is innervation an early target in autoimmune diabetes? Trends Immunol 2003. 24:574-579. [DOD]
- Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, Sampson A, Afifiyan F, Elford A, Jackowski G, Becker DJ, Santamaria P, Ohashi P, Dosch HM. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med 2003. 9:198-205. [DOD] [CrossRef]
- Lindsay JR, Nieman LK. The hypothalamic-pituitary-adrenal axis in pregnancy: challenges in disease detection and treatment. Endocr Rev 2005. 26:775-799. [DOD] [CrossRef]
- Thompson A, Arany EJ, Hill DJ, Yang K. Glucocorticoid receptor expression is altered in pancreatic beta cells of the non-obese diabetic mouse during postnatal development. Metabolism 2002. 51:765-768. [DOD] [CrossRef]
- Beauquis J, Saravia F, Coulaud J, Roig P, Dardenne M, Homo-Delarche F, De Nicola A. Prominently decreased hippocampal neurogenesis in a spontaneous model of type 1 diabetes, the nonobese diabetic mouse. Exp Neurol 2008. 210(2):359-367. [DOD] [CrossRef]
- Gaillard RC, Daneva T, Hadid R, Muller K, Spinedi E. The hypothalamo-pituitary-adrenal axis of athymic Swiss nude mice. The implications of T lymphocytes in the ACTH release from immune cells. Ann N Y Acad Sci 1998. 840:480-490. [DOD] [CrossRef]
- Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 1999. 5:601-604. [DOD]
- Zeidler A, Shargill NS, Chan TM. Peripheral insulin insensitivity in the hyperglycemic athymic nude mouse: similarity to noninsulin-dependent diabetes mellitus. Proc Soc Exp Biol Med 1991. 196:457-460. [DOD]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009. 15:930-939. [DOD] [CrossRef]
- Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 2009. 15:921-929. [DOD] [CrossRef]
- Chaparro RJ, Konigshofer Y, Beilhack GF, Shizuru JA, McDevitt HO, Chien YH. Nonobese diabetic mice express aspects of both type 1 and type 2 diabetes. Proc Natl Acad Sci U S A 2006. 103:12475-12480. [DOD] [CrossRef]
- Kagohashi Y, Udagawa J, Abiru N, Kobayashi M, Moriyama K, Otani H. Maternal factors in a model of type 1 diabetes differentially affect the development of insulitis and overt diabetes in offspring. Diabetes 2005. 54:2026-2031. [DOD] [CrossRef]
- Wilson SS, DeLuca D. NOD fetal thymus organ culture: an in vitro model for the development of T cells involved in IDDM. J Autoimmun 1997. 10:461-472. [DOD] [CrossRef]
- Senecat O, Martignat L, Elmansour A, Charbonnel B, Sai P. Diabetes enhancement and increased islet antigen expression following neonatal injections of glucose and arginine in non-obese diabetic mice. Metabolism 1994. 43:1410-1418. [DOD] [CrossRef]
- Saravia-Fernandez F, Durant S, el Hasnaoui A, Dardenne M, Homo-Delarche F. Environmental and experimental procedures leading to variations in the incidence of diabetes in the nonobese diabetic (NOD) mouse. Autoimmunity 1996. 24:113-121. [DOD] [CrossRef]
- Dahlquist G, Kallen B. Early neonatal events and the disease incidence in nonobese diabetic mice. Pediatr Res 1997. 42:489-491. [DOD] [CrossRef]
- Washburn LR, Dang H, Tian J, Kaufman DL. The postnatal maternal environment influences diabetes development in nonobese diabetic mice. J Autoimmun 2007. 28:19-23. [DOD] [CrossRef]
This article has been cited by other articles:

|
Type 1 diabetes in mice and men: gene expression profiling to investigate disease pathogenesis
Yip L, Fathman CG
Immunol Res 2014. 58(2-3):340-350
|
|

|
Diminished adenosine A1 receptor expression in pancreatic α-cells may contribute to the pathology of type 1 diabetes
Yip L, Taylor C, Whiting CC, Fathman CG
Diabetes 2013. 62(12):4208-4219
|
|

|
Loss of HGF/c-Met signaling in pancreatic β-cells leads to incomplete maternal β-cell adaptation and gestational diabetes mellitus
Demirci C, Ernst S, Alvarez-Perez JC, Rosa T, Valle S, Shridhar V, Casinelli GP, Alonso LC, Vasavada RC, García-Ocana A
Diabetes 2012. 61(5):1143-1152
|
|
|


)
)
)
)
)
)
)

