Review
| Rev Diabet Stud,
2011,
8(3):418-431 |
DOI 10.1900/RDS.2011.8.418 |
GLP-1, the Gut-Brain, and Brain-Periphery Axes
Cendrine Cabou, Remy Burcelin
INSERM (Institut National de la Sante et de la Recherche Medicale), U1048, Institute of Metabolic and Cardiovascular Diseases Rangueil, University of Toulouse III (Paul-Sabatier), (C.C, R.B), and the Faculty of Pharmacy, Toulouse, France
Address correspondence to: Remy Burcelin, e-mail: remy.burcelin@inserm.fr
Manuscript submitted June 30, 2011; resubmitted August 26, 2011; accepted September 5, 2011.
Keywords: type 2 diabetes, glycemic control, incretins, GLP-1, DPP-4, brain, exendin-4, glucose homeostasis, area postrema
Abstract
Glucagon-like peptide 1 (GLP-1) is a gut hormone which directly binds to the GLP-1 receptor located at the surface of the pancreatic β-cells to enhance glucose-induced insulin secretion. In addition to its pancreatic effects, GLP-1 can induce metabolic actions by interacting with its receptors expressed on nerve cells in the gut and the brain. GLP-1 can also be considered as a neuropeptide synthesized by neuronal cells in the brain stem that release the peptide directly into the hypothalamus. In this environment, GLP-1 is assumed to control numerous metabolic and cardiovascular functions such as insulin secretion, glucose production and utilization, and arterial blood flow. However, the exact roles of these two locations in the regulation of glucose homeostasis are not well understood. In this review, we highlight the latest experimental data supporting the role of the gut-brain and brain-periphery axes in the control of glucose homeostasis. We also focus our attention on the relevance of β-cell and brain cell targeting by gut GLP-1 for the regulation of glucose homeostasis. In addition to its action on β-cells, we find that understanding the physiological role of GLP-1 will help to develop GLP-1-based therapies to control glycemia in type 2 diabetes by triggering the gut-brain axis or the brain directly. This pleiotropic action of GLP-1 is an important concept that may help to explain the observation that, during their treatment, type 2 diabetic patients can be identified as 'responders' and 'non-responders'.
Abbreviations: 3drV - dorsal third ventricle; 3V - third ventricle; AMP - adenosine monophosphate; ANS - autonomic nervous system; AP - area postrema; ARC - arcuate; AgRP - agouti-related peptide; ATP - adenosine triphosphate; CART - cocaine- and amphetamine-regulated transcript; CC - central canal; cDNA - complementary deoxyribonucleic acid; DMN - dorsomedian; DMNX - dorsal motor nucleus of the vagus; DPP-4 - dipeptidyl peptidase-4; DVC - dorsal vagal complex; GIP - glucose-dependent insulinotropic polypeptide; GLP-1 - glucagon-like peptide-1; Glp1r - GLP-1 receptor; GLP-2 - glucagon-like peptide-2; GRPP - glycentin related polypeptide; HFD - high fat diet; icv - intracerebroventricular; IP-1 - intervening peptide 1; KATP - ATP-sensitive potassium; KO - knock-out; LV - lateral ventricle; MPF - major proglucagon fragment; mRNA - messenger ribonucleic acid; NEP - neutral endopeptidase; NPY - neuropeptide Y; NTS - solitary tract nucleus; PKA - protein kinases A; PKC - protein kinases C; POMC - propiomelanocortin; PVN - paraventricular
Introduction
Glucose-induced insulin secretion is regulated by several neural and hormonal stimuli. In particular, hormones secreted by intestinal endocrine cells potently stimulate glucose-induced insulin secretion after nutrient absorption. These hormones, called gluco-incretins or insulinotropic hormones, are major regulators of postprandial glucose homeostasis. The first incretin identified was called gastric inhibitory polypeptide (GIP) as it inhibits gastric acid secretion [1]. It was renamed glucose-dependent insulinotropic polypeptide to point to its incretin effect, thus better reflecting its physiological action. The second incretin to be discovered was called glucagon-like peptide 1 (GLP-1). Together, these incretins regulate the function of the endocrine pancreas and specific brain functions. Each incretin has its own specific action on the gastrointestinal tract, blood vessels, heart, adipose tissue, and liver. Altogether, incretins control body weight and glycemia. Consequently, the metabolic action of incretins has served as the basis for multiple strategies in the treatment of type 2 diabetes.
A considerable amount of data is now available regarding the insulin secretory effect of incretins and the efficacy of incretin-based therapies in type 2 diabetes. Most of the data focused on GLP-1. This is most likely because it is more suitable for the treatment of diabetes since it is not lipogenic compared to GIP. Several studies have shown that type 2 diabetic patients are resistant to GIP treatment. Also, over the course of the last decade, GLP-1 was shown to play an important role in the control of cardiovascular functions and the brain Some findings point to an indirect role for the control of glucose metabolism, which has led to revisiting the metabolic action of the incretins. Firstly, 80-90% of the secreted GLP-1 is degraded by the dipeptidyl-peptidase 4 (DPP-4) before reaching insulin secreting β-cells. Secondly, a gut-to-brain GLP-1- and glucose-dependent axis has been described involving the vagus nerve for the transmission of nutritional signals towards the brain. Thirdly, GLP-1 and the GLP-1 receptor are present in the brain. Finally, multiple physiological effects are associated with the role of incretins in the brain. Consequently, the central action of the incretins in type 2 diabetic humans is now a matter of debate. As most studies have been performed on GLP-1, this review addresses the effects of GLP-1 on the brain in the regulation of energy homeostasis. It is noteworthy that recent literature points to a neuroprotective role for GLP-1, which, although important, is beyond the scope of this review.
The incretin concept
The incretin concept emerged from the observation that oral administration of an extract of pig duodenal mucosa could reduce hyperglycemia in diabetic patients [2]. The authors proposed that a stimulation of the internal pancreatic secretion would be mediated by this extract. Later, the development of an insulin immunoassay proved that this effect was due to an insulinotropic action [3]. At the same time, Elrick and Mc Intyre found that the insulin response to an oral glucose load was much greater than the response measured after an intravenous infusion of an equivalent amount of glucose [4, 5]. Their findings led to the conclusion that intestinal factors called incretins control postprandial insulin release. Subsequently, the two peptides secreted by the intestine were found to be involved in increased insulin secretion following an oral glucose load, GLP-1 and GIP.
First evidence on the existence of GLP-1 was found in patients with small-bowel resection who had a decreased incretin activity, despite plasma GIP remaining at normal level [6]. Later, GLP-1 was discovered following the cloning and sequencing of mammalian proglucagon genes [7, 8]. GIP is secreted by enteroendocrine K-cells, found predominantly in the proximal gut (duodenum-jejunum) [9, 10]. GLP-1 secreting L-cells are mainly localized in the distal gut (ileum-colon) [11-12]. In healthy humans, the intravenous infusion of either of these two peptides contributes nearly equally to the incretin effect in the fasted state, or under an euglycemic clamp obtained with a glucose infusion [13]. However, during hyperglycemia GLP-1 is more insulinotropic than GIP. Importantly, in most cases, GLP-1 maintains its insulinotropic effect in diabetic patients, but not GIP [14-17].
Peripheral GLP-1: secretion, metabolism and insulinotropic action
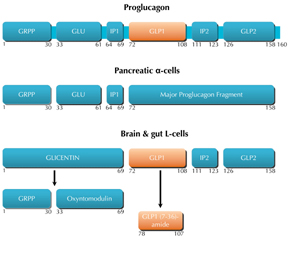
In the intestine, GLP-1 is synthesized by the pre-proglucagon gene (Figure 1), which is mainly expressed in intestinal enteroendocrine L-cells, and to a lesser extent in pancreatic α-cells and neurons. Although the mRNA transcript sequence is identical in these organs, it leads to the generation of a peptide precursor, namely proglucagon, which undergoes different proteolytic cleavages, and yields different end products in these tissues [11, 18-20] (Figure 1). In L-cells, the end products are GLP-1, GLP-2, glicentin, and oxyntomodulin. GLP-1 is first synthesized as an immature form with 37 amino acids. It is then cleaved and amidated at the C-terminal end to compose the major bioactive form of GLP-1: a peptide with 30 amino acids, called GLP-1 (7-36)-amide, which is found in the brain and circulating blood of humans and rodents [21, 22].
 |
 |
Figure 1. Schematic representation of the structure of proglucagon and its post-translational processing in different tissues. In the pancreas, post-translational processing of proglucagon leads to the generation of glycentin related polypeptide (GRPP), glucagon, intervening peptide (IP-1), and a major proglucagon fragment (MPF). In the brain and the gut, post-translational processing of proglucagon liberates GLP-1, GLP-2, IP-2, glicentin, and oxyntomodulin.. Proconvertases and carboxypeptidases are responsible for the processing of the mature peptides. Numbers represent the position of amino acids following the signal peptide (not shown). |
|
Oral glucose absorption induces GLP-1 (7-36)-amide release from intestinal L-cells into intestinal capillaries. It reaches the portal vein, then liver and pancreas, where it binds to a specific receptor expressed on pancreatic β-cells, and enhances glucose-induced insulin secretion. This glucose-dependent physiological mechanism is the basis of an important therapeutic advantage that avoids iatrogenic hypoglycemia. Thus, oral glucose is a potent stimulus for the release of GLP-1, and this does not occur when glucose bypasses the absorptive processes in the gut. The molecular events leading to GLP-1 release by L-cells have been described in a recent review [23]. GLP-1 also decreases glucagon secretion by pancreatic α-cells [14], and increases the insulin to glucagon ratio, which improves insulin sensitivity. Furthermore, GLP-1 increases β-cell mass by stimulating proliferation, inducing islet neogenesis, and inhibiting apoptosis [24-25]. Because of these potent insulinotropic actions, GLP-1 has attracted considerable attention as the major candidate for the role of the putative 'incretin' hormone.
DPP-4, which is ubiquitously found in the capillary endothelium, rapidly inactivates GLP-1 (7-36)-amide in the intestinal capillaries, the hepatoportal vein, and the periphery [26]. DPP-4 mediates GLP-1 degradation by cleaving the di-peptide at the N-terminus and removing the histidine-alanine dipeptide, yielding GLP-1 (9-36)-amide [27]. DPP-4 is present in the brain, where it shows high activity when measured in the hypothalamus, hippocampus, circumventricular organs, choroid plexus, and leptomeninges [28-29]. GLP-1(7-36)-amide is also degraded by another membrane-bound peptidase, called neutral endopeptidase 24.11 (NEP24.11), which is expressed in the periphery and the brain [30]. This peptidase degrades GLP-1 (7-36)-amide into smaller peptides including GLP-1 (28-36)-amide and GLP-1 (32-36)-amide [31]. Thus, after release from intestinal L-cells, the highest GLP-1 concentration occurs in the intestinal submucosal extracellular space, while intermediate levels can be found in the hepatoportal vein. Comparatively low concentrations are found in the systemic circulation [32]. It is estimated that less than 10% of intestinal GLP-1 is active after the first hepatic pass, and reaches the blood circulation to target peripheral organs such as the brain [32].
Because of its rapid inactivation by enzymes, GLP-1 (7-36)-amide has a very short half-life in the blood circulation, approximately 2 minutes in human and most mammalian species. It has been proposed that GLP-1 secretion in response to meal ingestion is reduced in type 2 diabetes [16, 33], although its relative insulinotropic activity is preserved [15]. However, a recent meta-analysis concluded that GLP-1 secretion in patients could be increased, normal, or reduced [34]. Therefore, debate is still ongoing, although long-lasting active analogues of GLP-1 and inhibitors of DPP4 have already been developed as drugs. These drugs are designed to increase the stability of the physiologically released hormone in type 2 diabetes [35]. At present, there is debate whether both drugs belong to the same therapeutic class.
Brain GLP-1 signaling and metabolic actions
Experimental studies carried out in rodent brains show that GLP-1 and its receptor are synthesized in selective brain areas, mainly in brainstem and hypothalamus. The localization of GLP-1 receptors in the brain may help us to understand the physiological role of brain GLP-1 signaling.
Brain GLP-1 synthesis
In the brain, pre-proglucagon expression is found in the solitary tract nucleus (NTS) and in the brainstem [20, 22]. Post-translational processing of proglucagon is similar to L-cell processing, and leads to GLP-1 with roughly equimolecular amounts of glicentin and oxyntomodulin [20, 22] (Figure 2). GLP-1 (7-36)-amide is synthesized by neuronal cell bodies in the caudal region of the NTS in the brainstem. There is a nucleus that forms the border of the caudal part of the fourth ventricle (Figure 3). The NTS is located in the dorsal vagal complex (DVC), which includes the area postrema (AP) and the dorsal motor nucleus of the vagus (DMNX). This complex receives visceral sensory inputs, generated by the vagal nerves that innervate the gastro-duodenal tract [36]. The distribution of cells expressing pre-proglucagon is not limited to a cluster of neurons in the caudal part of the NTS. It is also found in a small number of neurons that extend laterally from the NTS through the dorsal reticular area into the A1 area located in the medulla [20, 37-39].
 |
|
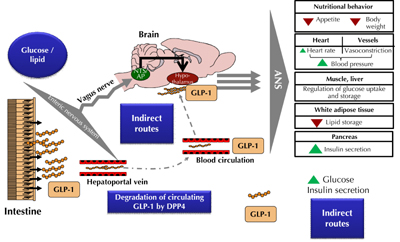
Figure 2. The gut-to-brain and the brain-to-periphery axes are part of GLP-1 metabolic and vascular functions. In response to glucose and lipid, GLP-1 is secreted by the intestine into the mesenteric capillaries and released into the hepatoportal vein. This activates termination ends from the vagus nerve to generate a neural signal towards the brain stem. The corresponding nuclei, such as the nucleus of the solitary tract nucleus (NTS) and the area postrema (AP), send axons to the hypothalamus, which release GLP-1 and activate the receptors. Then, a new signal is sent towards peripheral tissues through the autonomic nervous system (ANS) to regulate numerous functions. In blood and tissues, DPP-4 continuously degrades GLP-1. The remaining hormone could reach β-cells and enhance glucose-induced insulin secretion through the direct route, or by targeting the brain, through the indirect route. |
|
 |
|
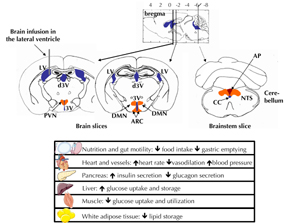
Figure 3. Schematic brain GLP-1 receptors localization in the mouse brain. GLP-1 receptors have been primarily identified in the paraventricular (PVN), dorsomedian (DMN), and arcuate (ARC) hypothalamic nuclei. In the brainstem, they were also localized in the nucleus of the tractus solitarius (NTS) and the area postrema (AP). These two last areas in the brainstem also contain the cell bodies of neurons synthesizing the GLP-1 which projects into the hypothalamus. All these localizations contribute to the metabolic effects described in the figure. Ventricles are represented in blue. CC: central canal. 3V: third ventricle. LV: lateral ventricle. 3drV: dorsal third ventricle. |
|
In retrograde tracing studies, it has been shown that GLP-1 immunoreactive fibers in the hypothalamus originate from NTS cell bodies [37]. These efferent projections from the NTS densely innervate the hypothalamus and mainly the paraventricular and the dorsomedial nuclei [20, 37, 39]. Moreover, moderate innervations are located in the arcuate nuclei and the subfornical organ [20]. Numerous GLP-1 immunoreactive fibers can also be found in extrahypothalamic areas such as thalamic and cortical areas. In the brainstem, fibers project towards reticular formation and spinal cord [40, 41].
Brain GLP-1 receptors
GLP-1 receptor cDNA from human [42] and rat [43] brains has been cloned and sequenced. The deduced amino acid sequences are the same as those found in pancreatic islets [44]. More precisely, both receptors are 463 amino acids in length, and show 90% amino acid sequence homology [44-46]. The receptor belongs to the class B family of 7 transmembrane-spanning G-protein-coupled receptors [47]. This receptor is linked to the adenylate cyclase system and protein kinase A (PKA) activation [48]. However, it has been shown that the receptor can also activate the phospholipase C pathway on pancreatic β-cells, which leads to protein kinases C (PKC) stimulation [46, 49, 50].
In situ hybridization studies with GLP-1 receptor mRNA and binding studies using radiolabeled GLP-1 have found labeled cells distributed throughout the entire brain in rodents [41, 51, 52] and humans [53]. These studies have also shown a high level of GLP-1 receptor-expressing neurons in the hypothalamus and the brainstem. These are two brain areas which are involved in the central control of energy homeostasis and autonomous functions. In the hypothalamus, the receptor is mainly located in the following nuclei: the arcuate nucleus, the paraventricular nucleus, the dorsomedial nucleus, and the supraotic nucleus (Figure 3). It can also be found in the DVC, especially in the NTS and the area postrema (Figure 3). Moreover, GLP-1 binding sites are present in the circumventricular organs such as the subfornical organ and the area postrema [41, 51, 54]. These last two locations could be the target for both peripheral GLP-1 of intestinal origin and GLP-1 synthesized in the nervous system.
The gut-to-brain GLP-1-dependent axis
The relatively low plasma level, and rapid metabolism of GLP-1 in the blood, raise questions about an alternative neural pathway that account for part of its endocrine effects on target organs such as pancreatic β-cells (especially on its effects on glucose tolerance). Thus, it has been suggested that GLP-1 secreted from L-cells could potentially influence brain neuronal activities via an alternative neural pathway initiated by sensors in the hepatic portal region [55] (Figure 2).
Indeed, experimental studies on rodents in our laboratory, and from others, have shown that glucose detection is associated with GLP-1 secretion and action on peripheral receptors localized on vagal nerve fibers in the enteric area [56], which includes the hepatoportal veins [55, 57-58]. Thereby, the vagus nerve transmits the metabolic information to the NTS in the brainstem [59-63], which relays the glucose signal to hypothalamic nuclei [59, 64]. The brain centralizes the metabolic information and generates new signals to guide the energetic flux towards tissues, either for energy use or storage. This process is called the gut-to-brain-to-periphery axis. Initially, we showed that a low rate infusion of glucose into the portal vein caused a paradoxical hypoglycemia [65-66]. This implied the activation of glucose sensors located in the portal vein, and the mechanism required the glucose transporter GLUT2 to detect glucose [67]. Hence, we suggested that mechanisms similar to those observed in insulin secreting β-cells were responsible for the activation of the gut-to-brain axis. Therefore, GLP-1 and somatostatin are hormones positively and negatively involved in the transmission of the nutritional signals towards the brain, respectively [65].
Another hypothesis is that circulating GLP-1 can access the brain to exert its metabolic effects, but this is still a matter of debate. Experimental studies in rodents have shown that circulating GLP-1, or its agonist exendin-4 (see below), can reach the brain as they can bind to blood-brain barrier-free circumventricular organs such as the subfornical organ close to the hypothalamus [68], and the area postrema in the brainstem [68-69]. Another mechanism determining central GLP-1 action could be GLP-1 neuronal secretion in the brain which appears in response to glucose detection by intestinal cells. In line with this observation, our laboratory found that a brain infusion of exendin-9, an antagonist of the GLP-1 receptor, can reduce muscle glucose utilization and its storage as glycogen in mice receiving glucose from an intragastric catheter [70]. However, the molecular mechanisms controlling this secretion remain unclear [71].
The physiologic role of GLP-1 receptors in the control of metabolic functions (Figure 2) has often been studied in rodents, following intracerebroventricular (icv) infusions of GLP-1 (7-36)-amide, or its analogues such as exendin-4 that are resistant to enzyme degradation (by NEP and DPP-4) [31]. Exendin-4, or exendin (4-39), is a natural peptide, purified from the salivary secretions of the Gila monster lizard (Heloderma suspectum). This peptide shows 53% sequence homology with the mammalian GLP-1 (7-36)-amide [72-73]. Binding studies using radiolabeled iodinated GLP-1 or exendin-4 have shown that both peptides specifically interact with the same receptor expressed on pancreatic β-cells isolated from rodents [73-74] and humans [44]. In the brain, these two peptides show an identical distribution pattern of binding sites [51]. Furthermore, these peptides increase the production of intracellular cyclic AMP and stimulate glucose-induced insulin secretion [44, 73-74]. All the abovementioned effects are inhibited by the GLP-1 receptor antagonist, exendin-9 or exendin (9-39) that is a truncated form of exendin-4 [44, 74]. The properties of exendin-4 as an agonist, and of exendin-9 as an antagonist, open the possibility of using these peptides as tools to study the physiology of the GLP-1 receptor.
Nutritional behavior and body weight
The anorectic properties of centrally administrated GLP-1 were described for the first time during the 1990's. These studies showed that a GLP-1 infusion into the brain ventricles of rodents inhibits short-term food and water intake, and decreases body weight in the long term [75-80]. These properties were also demonstrated in clinical studies: an intravenous GLP-1 infusion both decreased food intake and increased satiety in healthy [81], diabetic [82], and obese [83] subjects. Moreover, weight loss is reported in diabetic subjects treated for 6 weeks with GLP-1 administrated subcutaneously [84].
The mechanism by which GLP-1 inhibits food intake is not fully understood. It has been suggested that it could result from a central action of GLP-1. It has also been suggested that part of the anorectic effects of GLP-1 could result from its side effects such as nausea and inhibition of gastric emptying [85-87]. Another mechanism could be related to activation of peripheral GLP-1 receptors expressed by the vagus nerve [88-89], which relays metabolic signals to the brain. Ruttinmann et al. have shown that a peripheral injection of GLP-1, irrespective of its site of infusion (vena cava, intraperitoneal cavity, or portal vein), reduced food intake without affecting the subsequent inter-meal interval, the size of subsequent meals, or the cumulative food intake [88]. These effects were only attenuated when GLP-1 was intraperitoneally administered to rats with subdiaphragmatic vagal deafferentations. Furthermore, intraperitoneal injection of exenatide dose-dependently increases c-fos expression both in myenteric and submucosal neurons of the rat duodenum, but not in other locations like jejunum and ileum. Exenatide is a synthetic version of exendin-4 recently approved for clinical use in type 2 diabetic patients [90-92]. These peripheral effects are also associated with brain neuronal activations in the brainstem (area postrema, NTS, and dorsal motor nucleus of the vagus), and can be prevented by an intraperitoneal injection of exendin-9 [63]. Thus, the satiating effect of peripherally administered GLP-1 at least requires an activation of peripheral GLP-1 receptors, which metabolically transmits the signal of satiety to the brain using the vagus nerve. It is well demonstrated that GLP-1 can act on brain neuronal circuits in the hypothalamus involved in appetite control [77].
GLP-1 receptor mRNA is highly expressed in the hypothalamic arcuate nucleus, and precisely overlaps the area occupied by neurons co-expressing propiomelanocortin (POMC) [93]. Furthermore, icv infusion of GLP-1 in fasted rodents increases the synthesis of anorexigenic peptides (POMC and cocaine- and amphetamine-regulated transcript (CART)) both in arcuate and paraventricular nuclei [94]. In these brain locations, the peptide decreases the synthesis of orexigenic peptides (neuropeptide Y (NPY) and agouti-related peptide (AgRP)). We should note that in studies with GLP-1 injected in situ into rat brain hypothalamic nuclei the paraventricular nucleus [79], and not the arcuate nucleus [93], is involved in the anorectic response to GLP-1.
Cardiovascular effects
The first studies in the 1990's described insulin-induced vasodilatation of the femoral artery in humans, and its impact on the control of muscle glucose utilization and glucose tolerance [95-98]. Today, it is well known that the cardiovascular system plays a key role in regulating glucose homeostasis. In 1999, Barragan et al. demonstrated for the first time that brain GLP-1 infusion into the lateral ventricle of conscious rats increased blood pressure and heart rate in these rodents [99]. Later, these effects were replicated with a brain infusion of exendin-4, the agonist of the GLP-1 receptor [69, 100]. It is noteworthy that GLP-1 and exendin-4 both increase blood pressure and heart rate when they are infused into the blood in rats [54, 101], and that these effects can be prevented by a cerebral injection of exendin-9 [99]. This last observation suggests that circulating GLP-1 can regulate cardiovascular function by acting on the brain. We believe that the effect on blood pressure could be due to an indirect action on blood vessels. This hypothesis has been further demonstrated by our laboratory in healthy, conscious mice receiving an exendin-4 brain infusion in the lateral ventricle [102]. Our experimental work has shown that exendin-4 decreases insulin-induced vasodilatation of the femoral artery, and that this mechanism correlates with a decrease in insulin sensitivity.
It is well demonstrated that GLP-1 recruits the autonomic nervous system to modulate the cardiovascular tone [39]. Indeed, studies have reported on a role for the parasympathetic and sympathetic branch in this control mechanism. However, it is not clear which branch plays the more important role. Initially, experiments on healthy rodents demonstrated a potential role of the parasympathetic branch in the control of heart rate and blood pressure in response to peripheral or central GLP-1 action. This observation was demonstrated by vagotomy experiments [99] and pharmacological experiments using an icv injection of cholinergic tone inhibitors [100] in rodents receiving an exendin-4 brain infusion into the lateral ventricle. Other studies have assumed a role for the sympathetic tone. This has been suggested by an experiment showing that an intravenous or cerebral infusion of exendin-4 in rodents can stimulate c-fos expression (a marker of neuronal activation) in catecholaminergic neurons of the brainstem and in particular in the area postrema [54, 69]. Consequently, the area postrema has been thought to regulate these hemodynamic effects. In humans, a clinical study on healthy subjects assessed the effects of GLP-1 infusion on blood pressure and heart rate, and the possible activation of the sympathetic nervous system. The authors did not find any clear variation in systolic or diastolic blood pressure, but there was increased sympathetic nerve activity in skeletal muscles [103].
Cardiac effects have not been described in clinical studies with type 2 diabetic subjects. Although a 48 hour continuous subcutaneous GLP-1 infusion has no chronotropic effect, it can decrease both systolic and diastolic blood pressure in patients [104]. Interestingly, the same observations are reported in long-term studies (12 weeks to 6 months) with exenatide [105-106]. However, in non-diabetic patients with congestive heart failure, a minor increase in heart rate and diastolic blood pressure was observed during a 48 hour continuous subcutaneous GLP-1 infusion [107]. In conclusion, these observations suggest that the effects of GLP-1 on cardiovascular function seem to differ in clinical studies, and still remain to be unraveled. It is important to note that a recent review based on clinical trials has reported a positive beneficial impact of treatments with GLP-1, or its agonists (exenatide or liraglutide), on cardiac function in diabetic subjects. In part, these therapies show cardioprotective effects by reducing cardiovascular risk factors such as glycemia, lipidemia, blood pressure, and body weight [108].
Action on peripheral glucose homeostasis and fuel partitioning
Data from healthy rodents
Data from our laboratory, and from others, have shown that brain GLP-1 receptor signaling has metabolic effects on peripheral glucose tolerance. We were able to show in mice that intragastric low-rate glucose infusion (insufficient to affect systemic glucose levels) triggers skeletal muscle glucose uptake and glycogen synthesis, which highlights the significance of the gut-to-brain-to-periphery axis [70]. This effect is controlled by brain GLP-1 receptor activation, as it is abolished by prior icv administration of the GLP-1 receptor antagonist, exendin (9-39), and it is absent in GLP-1 receptor knock-out (KO) mice. We concluded that prandial perception of food facilitates a central GLP-1 receptor-dependent mechanism to improve glucose disposal. The quantification of the c-fos expression pattern in brainstem and hypothalamus showed that the enteric glucose sensor system sent signals to these brain areas. Neuronal cells in the NTS were activated by a low-rate intragastric glucose infusion, whereas cells from the arcuate nucleus of the hypothalamus were switched to rest [59]. These effects were attenuated in GLP-1 receptor KO mice, suggesting a role for brain GLP-1 signaling in influencing neural circuits to regulate glucose metabolism.
We have also demonstrated that brain GLP-1 signaling monitors the development of energy reserves in preparation for the fasting phase following a meal. We used hyperglycemic and hyperinsulinemic glucose clamps to raise systemic glucose and insulin concentrations to levels similar to those in prandial states. After exendin-4 activation of central GLP-1 receptors, we found reduced insulin-induced glucose uptake in skeletal muscles which favored hepatic glycogen stores [70]. In these experimental conditions, our results also showed that icv exendin-4 infusion increases insulin secretion by pancreatic β-cells, but prevents the vasodilatatory action of this hormone on the femoral artery [102]. We have further demonstrated that exendin-4 impairs muscle blood flow, an effect which contributes in part to the decline in skeletal muscle glucose utilization and muscle glucose uptake. During these experiments, we also found that icv brain infusion of exendin-4 activates neural pathways such as the vagus nerve [102] and muscle innervation [70], which mediates peripheral effects on muscle blood flow and insulin resistance. We assume that these physiological mechanisms prevent an overutilization of glucose by muscles during feeding to save glucose for the liver, and to prepare efficiently for the next fasting state [70]. However, the brain molecular and cellular mechanisms controlling the metabolic effects of brain GLP-1 remain unknown, and need to be investigated.
Sandoval et al. showed that the hypothalamic arcuate nucleus, and not the paraventricular nucleus, could be involved in this regulation. Indeed, the group showed that a GLP-1 infusion directly into the arcuate, and not the paraventricular nucleus, decreased glucose production and glucose uptake by peripheral tissues during clamp studies [93]. Also, co-infusion of a KATP channel blocker with GLP-1 into the arcuate nucleus inhibited the effect of GLP-1 on peripheral glucose metabolism [93]. These data confirm that central glucose-sensing neurons in the hypothalamic brain area are involved in brain GLP-1 signaling and control glucose tolerance. Finally, Nogueiras et al. demonstrated that a continuous icv infusion of GLP-1 in mice, directly and potently decreased lipid storage in white adipose tissue. This mechanism is independent of food intake and appears to be, in part, mediated by the sympathetic nervous system and the β-2 adrenoreceptors expressed in white adipose tissue [109].
In a recently published work, we have investigated the molecular mechanisms activated by GLP-1 in the brain that coordinate both vascular function and peripheral muscle glucose metabolism [110]. In vitro studies using rat pancreatic β-cells had previously shown that GLP-1 can activate PKC to modulate insulin secretion [50]. According to this observation, we support the hypothesis that these enzymes could be part of the molecular mechanisms of brain GLP-1 signaling. PKC are serine/threonine kinases that belong to intracellular signaling pathways. These enzymes can contribute to the regulation of synthesis and release of neurotransmitters, and to electrical activation of neuronal cells [111]. Therefore, we have repeated our experimental hyperglycemic and hyperinsulinemic clamp studies described above [102]. We observed that the brain exendin-4 infusion acutely triggers the translocation of cytoplasmic PKC-δ to the plasma membrane in cells from the hypothalamus. These functional and molecular effects were blocked by an icv brain exendin-9 infusion, or the genetic invalidation of the GLP-1 receptor (Glp1r-/- mice). Thus, our recent data have shown for the first time that brain GLP-1 can activate hypothalamic PKC-δ to decrease insulin-stimulated vasodilatation and whole body glucose utilization.
Data from diabetic and obese rodents
What is known about brain GLP-1 signaling during a state of overnutrition such as obesity or type 2 diabetes? Experimental studies on diabetic rodents have shown that brain GLP-1 signaling is stimulated during diabetes, and may derange glycemic control. This observation is supported by previous data from our group, and by others. It has been shown that expression of the proglucagon gene encoding GLP-1 is increased in the brainstem of diabetic high fat diet (HFD)-fed mice [112], and obese Zucker rats [38], which both contain cell bodies of GLP-1 neurons oriented towards the hypothalamus. Following this observation, and based on the hypothesis that GLP-1 can activate PKC, as demonstrated in pancreatic β-cells [50] or during an exendin-4 brain infusion, we have demonstrated that overall PKC activity is increased in the hypothalamus of diabetic HFD-fed mice in the fed state and during hyperinsulinemic euglycemic clamps [110]. This increased activity is associated with a translocation of both PKC-δ and -α to the plasma membrane of hypothalamic cells. However, exendin-9 brain infusion only inhibits PKC-δ translocation and improves the otherwise impaired insulin sensitivity and vasoconstriction observed in the control group.
Furthermore, the disruption of the GLP-1 receptor in Glp1r-/- mice prevents both overall PKC activation and PKC-δ translocation to the membrane fraction in healthy and diabetic Glp1r-/- mice. In this latter group, the genetic deletion seems to prevent both the impairment of the vascular blood flow and the insulin sensitivity during clamp studies. Thus, all these observations suggest that brain GLP-1 activity is increased during metabolic diseases. We believe that this overactivity might be due to an insidious increase in glycemia, which chronically stimulates the basal activity of brain neuronal cells. Therefore, this central mechanism could alter the metabolic effects of hormones released during the postprandial state, such as insulin.
Conclusions
Brain GLP-1 action has been studied over the last decade, but its physiological impact is still not completely understood. Numerous studies reported in the literature indicate a common effect of both peripheral and central GLP-1 receptor signaling on glucose homeostasis to promote glucose tolerance. These observations point to a cross-talk between circulating GLP-1 and the brain, but the relative importance of peripheral versus central GLP-1 for the control of glucose homeostasis is not known.
We think that the peripheral action of GLP-1 may play a critical role in glycemic regulation, perhaps even more important than its central action, because it initiates the metabolic information which is then redirected towards peripheral organs by the brain. It is also possible that the peripheral secretion indirectly regulates the level of activation of brain neuronal circuits expressing the GLP-1 receptor. We raise this hypothesis from observations obtained in mice invalidated for the GLP-1 receptor. Our recent data show that these mice do not develop the metabolic phenotype which was very similar to that observed in the group of diabetic mice brain-infused with the GLP-1 receptor antagonist, exendin-9 [110]. This observation suggests that the peripheral receptor may play a critical role in diabetes development.
It is also important to find out whether long-acting GLP-1 analogs could improve glucose tolerance in type 2 diabetes by acting on the brain. A similar conceptual question could be applied to DPP-4 inhibitors. As opposed to GLP-1 analogues, DPP-4 inhibitor treatment increases portal vein GLP-1 concentrations, and activates the gut-to-brain vagus nerve activity [113].
Hence, the identification of the gut-to-brain-to-periphery axis opens new avenues for the treatment of type 2 diabetes. The high concentration of circulating GLP-1 analogue obtained during the treatment of type 2 diabetes could reach the brain to favor neuroprotection and control of food intake. It is noteworthy that the effect of brain GLP-1 on insulin action and vascular blood flow is clearly analyzed in animals, but have never been mentioned in relation to humans. We consider that there is a strong case for such studies in humans.
Beside GLP-1 analogues, the use of DPP4 inhibitors could offer different therapeutic strategies. We can be sure that they recruit the gut-to-brain vagus nerve-dependent axis, which is unlikely the case for GLP-1 analogs. Different patient conditions could be treated, and therapies could be combined where appropriate. Such new incretin-based therapies, utilizing newly discovered modes of action, would need clinical trials to validate them, but have a great potential for a new era of diabetes treatment.
Disclosures: The authors report no conflict of interests.
Acknowledgments:
We would like to thank John Woodley for language editing.
References
- Brown JC, Dryburgh JR, Ross SA, Dupre J. Identification and actions of gastric inhibitory polypeptide. Recent Prog Horm Res 1975. 31:487-532. [DOD]
- Moore B. On the treatment of Diabetus mellitus by acid extract of Duodenal Mucous Membrane. Biochem J 1906. 1:28-38. [DOD]
- Dupre J, Beck JC. Stimulation of release of insulin by an extract of intestinal mucosa. Diabetes 1966. 15:555-559. [DOD]
- Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab 1964. 24:1076-1082. [DOD] [CrossRef]
- McIntyre N, Holdsworth CD, Turner DS. Intestinal factors in the control of insulin secretion. J Clin Endocrinol Metab 1965. 25:1317-1324. [DOD] [CrossRef]
- Lauritsen KB, Moody AJ, Christensen KC, Lindkaer Jensen S. Gastric inhibitory polypeptide (GIP) and insulin release after small-bowel resection in man. Scand J Gastroenterol 1980. 15:833-840. [DOD] [CrossRef]
- Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest 1987. 79:616-619. [DOD] [CrossRef]
- Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 1987. 2:1300-1304. [DOD] [CrossRef]
- Polak JM, Bloom SR, Kuzio M, Brown JC, Pearse AG. Cellular localization of gastric inhibitory polypeptide in the duodenum and jejunum. Gut 1973. 14:284-288. [DOD] [CrossRef]
- Buffa R, Polak JM, Pearse AG, Solcia E, Grimelius L, Capella C. Identification of the intestinal cell storing gastric inhibitory peptide. Histochemistry 1975. 43:249-255. [DOD] [CrossRef]
- Orskov C, Holst JJ, Knuhtsen S, Baldissera FG, Poulsen SS, Nielsen OV. Glucagon-like peptides GLP-1 and GLP-2, predicted products of the glucagon gene, are secreted separately from pig small intestine but not pancreas. Endocrinology 1986. 119:1467-1475. [DOD] [CrossRef]
- Hoyt EC, Lund PK, Winesett DE, Fuller CR, Ghatei MA, Bloom SR, Ulshen MH. Effects of fasting, refeeding, and intraluminal triglyceride on proglucagon expression in jejunum and ileum. Diabetes 1996. 45:434-439. [DOD] [CrossRef]
- Vilsboll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 2003. 114:115-121. [DOD] [CrossRef]
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 (7-36 amide) but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993. 91:301-307. [DOD] [CrossRef]
- Elahi D, McAloon-Dyke M, Fukagawa NK, Meneilly GS, Sclater AL, Minaker KL, Habener JF, Andersen DK. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7-37) in normal and diabetic subjects. Regul Pept 1994. 51:63-74. [DOD] [CrossRef]
- Kjems LL, Holst JJ, Volund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 2003. 52:380-386. [DOD] [CrossRef]
- Mentis N, Vardarli I, Kothe LD, Holst JJ, Deacon CF, Theodorakis M, Meier JJ, Nauck MA. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 2011. 60:1270-1276. [DOD] [CrossRef]
- Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem 1986. 261:11880-11889. [DOD]
- Philippe J, Mojsov S, Drucker DJ, Habener JF. Proglucagon processing in a rat islet cell line resembles phenotype of intestine rather than pancreas. Endocrinology 1986. 119:2833-2839. [DOD] [CrossRef]
- Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience 1997. 77:257-270. [DOD] [CrossRef]
- Orskov C, Rabenhoj L, Wettergren A, Kofod H, Holst JJ. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 1994. 43:535-539. [DOD] [CrossRef]
- Kreymann B, Ghatei MA, Burnet P, Williams G, Kanse S, Diani AR, Bloom SR. Characterization of glucagon-like peptide-1-(7-36)amide in the hypothalamus. Brain Res 1989. 502:325-331. [DOD] [CrossRef]
- Reimann F. Molecular mechanisms underlying nutrient detection by incretin-secreting cells. Int Dairy J 2010. 20:236-242. [DOD] [CrossRef]
- Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem 2003. 278:471-478. [DOD] [CrossRef]
- Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, Bertolotto C, Di Mario U, Harlan DM, Perfetti R. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003. 144:5149-5158. [DOD] [CrossRef]
- Matheeussen V, Baerts L, De Meyer G, De Keulenaer G, Van der Veken P, Augustyns K, Dubois V, Scharpe S, De Meester I. Expression and spatial heterogeneity of dipeptidyl peptidases in endothelial cells of conduct vessels and capillaries. Biol Chem 2011. 392(3):189-198. [DOD] [CrossRef]
- Deacon CF. Circulation and degradation of GIP and GLP-1. Horm Metab Res 2004. 36:761-765. [DOD] [CrossRef]
- Alponti RF, Frezzatti R, Barone JM, Alegre Vde S, Silveira PF. Dipeptidyl peptidase IV in the hypothalamus and hippocampus of monosodium glutamate obese and food-deprived rats. Metabolism 2011. 60:234-242. [DOD] [CrossRef]
- Vrang N, Larsen PJ. Preproglucagon derived peptides GLP-1, GLP-2 and oxyntomodulin in the CNS: role of peripherally secreted and centrally produced peptides. Prog Neurobiol 2010. 92:442-462. [DOD] [CrossRef]
- Plamboeck A, Holst JJ, Carr RD, Deacon CF. Neutral endopeptidase 24.11 and dipeptidyl peptidase IV are both mediators of the degradation of glucagon-like peptide 1 in the anaesthetised pig. Diabetologia 2005. 48:1882-1890. [DOD] [CrossRef]
- Hupe-Sodmann K, McGregor GP, Bridenbaugh R, Goke R, Goke B, Thole H, Zimmermann B, Voigt K. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7-36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul Pept 1995. 58:149-156. [DOD] [CrossRef]
- Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-IV inhibitors. Diabetologia 2005. 48:612-615. [DOD] [CrossRef]
- Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, Holst JJ. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001. 86:3717-3723. [DOD] [CrossRef]
- Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia 2011. 54:10-18. [DOD]
- Green BD, Gault VA, O'Harte F P, Flatt PR. Structurally modified analogues of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) as future antidiabetic agents. Curr Pharm Des 2004. 10:3651-3662. [DOD] [CrossRef]
- Grill HJ. Leptin and the systems neuroscience of meal size control. Front Neuroendocrinol 2010. 31:61-78. [DOD] [CrossRef]
- Vrang N, Hansen M, Larsen PJ, Tang-Christensen M. Characterization of brainstem preproglucagon projections to the paraventricular and dorsomedial hypothalamic nuclei. Brain Res 2007. 1149:118-126. [DOD] [CrossRef]
- Vrang N, Larsen PJ, Jensen PB, Lykkegaard K, Artmann A, Larsen LK, Tang-Christensen M. Upregulation of the brainstem preproglucagon system in the obese Zucker rat. Brain Res 2008. 1187:116-124. [DOD] [CrossRef]
- Llewellyn-Smith IJ, Reimann F, Gribble FM, Trapp S. Preproglucagon neurons project widely to autonomic control areas in the mouse brain. Neuroscience 2011. 180:111-121. [DOD] [CrossRef]
- Jin SL, Han VK, Simmons JG, Towle AC, Lauder JM, Lund PK. Distribution of glucagonlike peptide I (GLP-I), glucagon, and glicentin in the rat brain: an immunocytochemical study. J Comp Neurol 1988. 271:519-532. [DOD] [CrossRef]
- Merchenthaler I, Lane M, Shughrue P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J Comp Neurol 1999. 403:261-280. [DOD] [CrossRef]
- Wei Y, Mojsov S. Tissue-specific expression of the human receptor for glucagon-like peptide-I: brain, heart and pancreatic forms have the same deduced amino acid sequences. FEBS Lett 1995. 358:219-224. [DOD] [CrossRef]
- Alvarez E, Roncero I, Chowen JA, Thorens B, Blazquez E. Expression of the glucagon-like peptide-1 receptor gene in rat brain. J Neurochem 1996. 66:920-927. [DOD] [CrossRef]
- Thorens B, Porret A, Buhler L, Deng SP, Morel P, Widmann C. Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9-39) an antagonist of the receptor. Diabetes 1993. 42:1678-1682. [DOD] [CrossRef]
- Thorens B. Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci U S A 1992. 89:8641-8645. [DOD] [CrossRef]
- Dillon JS, Tanizawa Y, Wheeler MB, Leng XH, Ligon BB, Rabin DU, Yoo-Warren H, Permutt MA, Boyd AE 3rd. Cloning and functional expression of the human glucagon-like peptide-1 (GLP-1) receptor. Endocrinology 1993. 133:1907-1910. [DOD] [CrossRef]
- Mayo KE, Miller LJ, Bataille D, Dalle S, Goke B, Thorens B, Drucker DJ. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev 2003. 55:167-194. [DOD] [CrossRef]
- Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U S A 1987. 84:3434-3438. [DOD] [CrossRef]
- Wheeler MB, Lu M, Dillon JS, Leng XH, Chen C, Boyd AE 3rd. Functional expression of the rat glucagon-like peptide-I receptor, evidence for coupling to both adenylyl cyclase and phospholipase-C. Endocrinology 1993. 133:57-62. [DOD] [CrossRef]
- Suzuki Y, Zhang H, Saito N, Kojima I, Urano T, Mogami H. Glucagon-like peptide 1 activates protein kinase C through Ca2+-dependent activation of phospholipase C in insulin-secreting cells. J Biol Chem 2006. 281:28499-28507. [DOD] [CrossRef]
- Goke R, Larsen PJ, Mikkelsen JD, Sheikh SP. Distribution of GLP-1 binding sites in the rat brain: evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur J Neurosci 1995. 7:2294-2300. [DOD] [CrossRef]
- Shughrue PJ, Lane MV, Merchenthaler I. Glucagon-like peptide-1 receptor (GLP1-R) mRNA in the rat hypothalamus. Endocrinology 1996. 137:5159-5162. [DOD] [CrossRef]
- Alvarez E, Martinez MD, Roncero I, Chowen JA, Garcia-Cuartero B, Gispert JD, Sanz C, Vazquez P, Maldonado A, de Caceres J, et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J Neurochem 2005. 92:798-806. [DOD] [CrossRef]
- Yamamoto H, Kishi T, Lee CE, Choi BJ, Fang H, Hollenberg AN, Drucker DJ, Elmquist JK. Glucagon-like peptide-1-responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. J Neurosci 2003. 23:2939-2946. [DOD]
- Burcelin R, Da Costa A, Drucker D, Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 2001. 50:1720-1728. [DOD] [CrossRef]
- Nakagawa A, Satake H, Nakabayashi H, Nishizawa M, Furuya K, Nakano S, Kigoshi T, Nakayama K, Uchida K. Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton Neurosci 2004. 110:36-43. [DOD] [CrossRef]
- Balkan B, Li X. Portal GLP-1 administration in rats augments the insulin response to glucose via neuronal mechanisms. Am J Physiol Regul Integr Comp Physiol 2000. 279:R1449-R1454. [DOD]
- Vahl TP, Tauchi M, Durler TS, Elfers EE, Fernandes TM, Bitner RD, Ellis KS, Woods SC, Seeley RJ, Herman JP, et al. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology 2007. 148:4965-4973. [DOD] [CrossRef]
- Knauf C, Cani PD, Kim DH, Iglesias MA, Chabo C, Waget A, Colom A, Rastrelli S, Delzenne NM, Drucker DJ, et al. Role of central nervous system glucagon-like Peptide-1 receptors in enteric glucose sensing. Diabetes 2008. 57:2603-2612. [DOD] [CrossRef]
- Adachi A. Projection of the hepatic vagal nerve in the medulla oblongata. J Auton Nerv Syst 1984. 10:287-293. [DOD] [CrossRef]
- Adachi A, Shimizu N, Oomura Y, Kobashi M. Convergence of hepatoportal glucose-sensitive afferent signals to glucose-sensitive units within the nucleus of the solitary tract. Neurosci Lett 1984. 46:215-218. [DOD] [CrossRef]
- Baumgartner I, Pacheco-Lopez G, Ruttimann EB, Arnold M, Asarian L, Langhans W, Geary N, Hillebrand JJ. Hepatic-portal vein infusions of glucagon-like peptide-1 reduce meal size and increase c-Fos expression in the nucleus tractus solitarii, area postrema and central nucleus of the amygdala in rats. J Neuroendocrinol 2010. 22:557-563. [DOD] [CrossRef]
- Washington MC, Raboin SJ, Thompson W, Larsen CJ, Sayegh AI. Exenatide reduces food intake and activates the enteric nervous system of the gastrointestinal tract and the dorsal vagal complex of the hindbrain in the rat by a GLP-1 receptor. Brain Res 2010. 1344:124-133. [DOD] [CrossRef]
- Shimizu N, Oomura Y, Novin D, Grijalva CV, Cooper PH. Functional correlations between lateral hypothalamic glucose-sensitive neurons and hepatic portal glucose-sensitive units in rat. Brain Res 1983. 265:49-54. [DOD] [CrossRef]
- Burcelin R, Dolci W, Thorens B. Portal glucose infusion in the mouse induces hypoglycemia: evidence that the hepatoportal glucose sensor stimulates glucose utilization. Diabetes 2000. 49:1635-1642. [DOD] [CrossRef]
- Burcelin R, Crivelli V, Perrin C, Da Costa A, Mu J, Kahn BB, Birnbaum MJ, Kahn CR, Vollenweider P, Thorens B. GLUT4, AMP kinase, but not the insulin receptor, are required for hepatoportal glucose sensor-stimulated muscle glucose utilization. J Clin Invest 2003. 111:1555-1562. [DOD]
- Burcelin R, Dolci W, Thorens B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes 2000. 49:1643-1648. [DOD] [CrossRef]
- Orskov C, Poulsen SS, Moller M, Holst JJ. Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide I. Diabetes 1996. 45:832-835. [DOD] [CrossRef]
- Yamamoto H, Lee CE, Marcus JN, Williams TD, Overton JM, Lopez ME, Hollenberg AN, Baggio L, Saper CB, Drucker DJ, et al. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. J Clin Invest 2002. 110:43-52. [DOD]
- Knauf C, Cani PD, Perrin C, Iglesias MA, Maury JF, Bernard E, Benhamed F, Gremeaux T, Drucker DJ, Kahn CR, et al. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Invest 2005. 115:3554-3563. [DOD] [CrossRef]
- Burcelin R. What is known, new and controversial about GLP-1? Minutes of the 1st European GLP-1 Club Meeting, Marseille, 28-29 May 2008. Diabetes Metab 2008. 34:627-630. [DOD] [CrossRef]
- Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem 1992. 267:7402-7405. [DOD]
- Raufman JP, Singh L, Singh G, Eng J. Truncated glucagon-like peptide-1 interacts with exendin receptors on dispersed acini from guinea pig pancreas. Identification of a mammalian analogue of the reptilian peptide exendin-4. J Biol Chem 1992. 267:21432-21437. [DOD]
- Goke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, Goke B. Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide 1-(7-36)-amide receptor of insulin-secreting beta-cells. J Biol Chem 1993. 268:19650-19655. [DOD]
- Navarro M, Rodriquez de Fonseca F, Alvarez E, Chowen JA, Zueco JA, Gomez R, Eng J, Blazquez E. Colocalization of glucagon-like peptide-1 (GLP-1) receptors, glucose transporter GLUT-2, and glucokinase mRNAs in rat hypothalamic cells: evidence for a role of GLP-1 receptor agonists as an inhibitory signal for food and water intake. J Neurochem 1996. 67:1982-1991. [DOD] [CrossRef]
- Scrocchi LA, Brown TJ, MaClusky N, Brubaker PL, Auerbach AB, Joyner AL, Drucker DJ. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med 1996. 2:1254-1258. [DOD] [CrossRef]
- Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, Choi SJ, Taylor GM, Heath MM, Lambert PD, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 1996. 379:69-72. [DOD] [CrossRef]
- Tang-Christensen M, Larsen PJ, Goke R, Fink-Jensen A, Jessop DS, Moller M, Sheikh SP. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol 1996. 271:R848-R856. [DOD]
- McMahon LR, Wellman PJ. PVN infusion of GLP-1-(7-36) amide suppresses feeding but does not induce aversion or alter locomotion in rats. Am J Physiol 1998. 274:R23-R29. [DOD]
- Meeran K, O'Shea D, Edwards CM, Turton MD, Heath MM, Gunn I, Abusnana S, Rossi M, Small CJ, et al. Repeated intracerebroventricular administration of glucagon-like peptide-1-(7-36) amide or exendin-(9-39) alters body weight in the rat. Endocrinology 1999. 140:244-250. [DOD] [CrossRef]
- Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest 1998. 101:515-520. [DOD] [CrossRef]
- Gutzwiller JP, Drewe J, Goke B, Schmidt H, Rohrer B, Lareida J, Beglinger C. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol 1999. 276:R1541-R1544. [DOD]
- Naslund E, Barkeling B, King N, Gutniak M, Blundell JE, Holst JJ, Rossner S, Hellstrom PM. Energy intake and appetite are suppressed by glucagon-like peptide-1 (GLP-1) in obese men. Int J Obes Relat Metab Disord 1999. 23:304-311. [DOD] [CrossRef]
- Meier JJ, Gallwitz B, Schmidt WE, Nauck MA. Glucagon-like peptide 1 as a regulator of food intake and body weight: therapeutic perspectives. Eur J Pharmacol 2002. 440:269-279. [DOD] [CrossRef]
- Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7-36) amide in type 2 (noninsulin-dependent) diabetic patients. J Clin Endocrinol Metab 1996. 81:327-332. [DOD] [CrossRef]
- Naslund E, Bogefors J, Skogar S, Gryback P, Jacobsson H, Holst JJ, Hellstrom PM. GLP-1 slows solid gastric emptying and inhibits insulin, glucagon, and PYY release in humans. Am J Physiol 1999. 277:R910-R916. [DOD]
- Verdich C, Flint A, Gutzwiller JP, Naslund E, Beglinger C, Hellstrom PM, Long SJ, Morgan LM, Holst JJ, Astrup A. A meta-analysis of the effect of glucagon-like peptide-1 (7-36) amide on ad libitum energy intake in humans. J Clin Endocrinol Metab 2001. 86:4382-4389. [DOD] [CrossRef]
- Ruttimann EB, Arnold M, Hillebrand JJ, Geary N, Langhans W. Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology 2009. 150:1174-1181. [DOD] [CrossRef]
- Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 2009. 150:1680-1687. [DOD] [CrossRef]
- Bergenstal RM, Wysham C, Macconell L, Malloy J, Walsh B, Yan P, Wilhelm K, Malone J, Porter LE. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION-2): a randomised trial. Lancet 2010. 376:431-439. [DOD] [CrossRef]
- Diamant M, Van Gaal L, Stranks S, Northrup J, Cao D, Taylor K, Trautmann M. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION-3): an open-label randomised trial. Lancet 2010. 375:2234-2243. [DOD] [CrossRef]
- Nielsen LL, Young AA, Parkes DG. Pharmacology of exenatide (synthetic exendin-4): a potential therapeutic for improved glycemic control of type 2 diabetes. Regul Pept 2004. 117:77-88. [DOD] [CrossRef]
- Sandoval DA, Bagnol D, Woods SC, D'Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 2008. 57:2046-2054. [DOD] [CrossRef]
- Seo S, Ju S, Chung H, Lee D, Park S. Acute effects of glucagon-like peptide-1 on hypothalamic neuropeptide and AMP activated kinase expression in fasted rats. Endocr J 2008. 55:867-874. [DOD] [CrossRef]
- Baron AD, Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G. Insulin-mediated skeletal muscle vasodilation contributes to both insulin sensitivity and responsiveness in lean humans. J Clin Invest 1995. 96:786-792. [DOD] [CrossRef]
- Baron AD, Laakso M, Brechtel G, Edelman SV. Mechanism of insulin resistance in insulin-dependent diabetes mellitus: a major role for reduced skeletal muscle blood flow. J Clin Endocrinol Metab 1991. 73:637-643. [DOD] [CrossRef]
- Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest 1996. 97:2601-2610. [DOD] [CrossRef]
- Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest 1994. 94:1172-1179. [DOD] [CrossRef]
- Barragan JM, Eng J, Rodriguez R, Blazquez E. Neural contribution to the effect of glucagon-like peptide-1-(7-36) amide on arterial blood pressure in rats. Am J Physiol 1999. 277:E784-E791. [DOD]
- Isbil-Buyukcoskun N, Gulec G. Effects of intracerebroventricularly injected glucagon-like peptide-1 on cardiovascular parameters; role of central cholinergic system and vasopressin. Regul Pept 2004. 118:33-38. [DOD] [CrossRef]
- Barragan JM, Rodriguez RE, Eng J, Blazquez E. Interactions of exendin-(9-39) with the effects of glucagon-like peptide-1-(7-36) amide and of exendin-4 on arterial blood pressure and heart rate in rats. Regul Pept 1996. 67:63-68. [DOD] [CrossRef]
- Cabou C, Campistron G, Marsollier N, Leloup C, Cruciani-Guglielmacci C, Penicaud L, Drucker DJ, Magnan C, Burcelin R. Brain glucagon-like peptide-1 regulates arterial blood flow, heart rate, and insulin sensitivity. Diabetes 2008. 57:2577-2587. [DOD] [CrossRef]
- Bharucha AE, Charkoudian N, Andrews CN, Camilleri M, Sletten D, Zinsmeister AR, Low PA. Effects of glucagon-like peptide-1, yohimbine, and nitrergic modulation on sympathetic and parasympathetic activity in humans. Am J Physiol Regul Integr Comp Physiol 2008. 295:R874-R880. [DOD] [CrossRef]
- Toft-Nielsen MB, Madsbad S, Holst JJ. Continuous subcutaneous infusion of glucagon-like peptide 1 lowers plasma glucose and reduces appetite in type 2 diabetic patients. Diabetes Care 1999. 22:1137-1143. [DOD] [CrossRef]
- Gill A, Hoogwerf BJ, Burger J, Bruce S, Macconell L, Yan P, Braun D, Giaconia J, Malone J. Effect of exenatide on heart rate and blood pressure in subjects with type 2 diabetes mellitus: a double-blind, placebo-controlled, randomized pilot study. Cardiovasc Diabetol 2010. 9:6. [DOD] [CrossRef]
- Okerson T, Yan P, Stonehouse A, Brodows R. Effects of exenatide on systolic blood pressure in subjects with type 2 diabetes. Am J Hypertens 2010. 23:334-339. [DOD] [CrossRef]
- Halbirk M, Norrelund H, Moller N, Holst JJ, Schmitz O, Nielsen R, Nielsen-Kudsk JE, Nielsen SS, Nielsen TT, Eiskjaer H, et al. Cardiovascular and metabolic effects of 48-h glucagon-like peptide-1 infusion in compensated chronic patients with heart failure. Am J Physiol Heart Circ Physiol 2010. 298:H1096-H1102. [DOD] [CrossRef]
- Verge D, Lopez X. Impact of GLP-1 and GLP-1 receptor agonists on cardiovascular risk factors in type 2 diabetes. Curr Diabetes Rev 2010. 6:191-200. [DOD] [CrossRef]
- Nogueiras R, Perez-Tilve D, Veyrat-Durebex C, Morgan DA, Varela L, Haynes WG, Patterson JT, Disse E, Pfluger PT, Lopez M, et al. Direct control of peripheral lipid deposition by CNS GLP-1 receptor signaling is mediated by the sympathetic nervous system and blunted in diet-induced obesity. J Neurosci 2009. 29:5916-5925. [DOD] [CrossRef]
- Cabou C, Vachoux C, Campistron G, Drucker DJ, Burcelin R. Brain GLP-1 signaling regulates femoral artery blood flow and insulin sensitivity through hypothalamic PKC-delta. Diabetes 2011. 60(9):2245-2256. [DOD] [CrossRef]
- Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci 1994. 17:551-567. [DOD] [CrossRef]
- Knauf C, Cani PD, Ait-Belgnaoui A, Benani A, Dray C, Cabou C, Colom A, Uldry M, Rastrelli S, Sabatier E, et al. Brain glucagon-like peptide 1 signaling controls the onset of high-fat diet-induced insulin resistance and reduces energy expenditure. Endocrinology 2008. 149:4768-4777. [DOD] [CrossRef]
- Waget A, Cabou C, Masseboeuf M, Cattan P, Armanet M, Karaca M, Castel J, Garret C, Payros G, Maida A, et al. Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology 2011. 152(8):3018-3029. [DOD] [CrossRef]
This article has been cited by other articles:

|
Procyanidins and their healthy protective effects against type 2 diabetes
Gonzalez-Abuin N, Pinent M, Casanova-Marti A, Arola L, Blay M, Ardevol A
Curr Med Chem 2015. 22(1):39-50
|
|

|
Central Nervous System Regulation of Intestinal Lipoprotein Metabolism by Glucagon-Like Peptide-1 via a Brain-Gut Axis
Farr S, Baker C, Naples M, Taher J, Iqbal J, Hussain M, Adeli K
Arterioscler Thromb Vasc Biol 2015. In press
|
|

|
Nutraceutical oleuropein supplementation prevents high fat diet-induced adiposity in mice
van der Stelt I, Hoek-van den Hil EF, Swarts HJ, Vervoort JJ, Hoving L, Skaltsounis L, Lemonakis N, Andreadou I, van Schothorst EM, Keijer J
J Funct Food 2015. 14:702-715
|
|

|
Activation of spinal glucagon-like peptide-1 receptors specifically suppresses pain hypersensitivity
Gong N, Xiao Q, Zhu B, Zhang CY, Wang YC, Fan H, Ma AN, Wang YX
J Neurosci 2014. 34(15):5322-5334
|
|

|
Incretin-based therapy and pancreatic beta cells
Chon S, Riveline JP, Blondeau B, Gautier JF
Diabetes Metab 2014. 40(6):411-422
|
|

|
A randomized dose-finding study demonstrating the efficacy and tolerability of albiglutide in Japanese patients with type 2 diabetes mellitus
Seino Y, Inagaki N, Miyahara H, Okuda I, Bush M, Ye J, Holland MC, Johnson S, Lewis E, Nakajima H
Curr Med Res Opin 2014. 30(6):1095-1106
|
|

|
Grape-seed procyanidins prevent the cafeteria-diet-induced decrease of glucagon-like peptide-1 production
Gonzalez-Abuin N, Martinez-Micaelo N, Blay M, Ardevol A, Pinent M
J Agric Food Chem 2014. 62(5):1066-1072
|
|

|
Glutathione peroxidase mimic ebselen improves glucose-stimulated insulin secretion in murine islets
Wang X, Yun JW, Lei XG
Antioxid Redox Signal 2014. 20(2):191-203
|
|

|
GLP-1-based strategies: a physiological analysis of differential mode of action
Burcelin R, Gourdy P, Dalle S
Physiology (Bethesda) 2014. 29(2):108-121
|
|

|
Linagliptin enhances neural stem cell proliferation after stroke in type 2 diabetic mice
Darsalia V, Olverling A, Larsson M, Mansouri S, Nathanson D, Nyström T, Klein T, Sjöholm A, Patrone C
Regul Pept 2014. 190-191:25-31
|
|

|
Regulation of glucose homeostasis by GLP-1
Nadkarni P, Chepurny OG, Holz GG
Prog Mol Biol Transl Sci 2014. 121:23-65
|
|

|
Physiological and pharmacological features of GLP-1 receptor agonists in type 2 diabetes
Burcelin R, Bertolini M
Med Malad Metab 2013. 7(4):331-339
|
|

|
GLP-1 effects on islets: hormonal, neuronal, or paracrine?
Donath MY, Burcelin R
Diabetes Care 2013. 36(Suppl 2):S145-S148
|
|

|
Incretins: what is known, new and controversial in 2013?
Burcelin R, Thorens B, European Club for the study of GLP-1 (EuCSGLP-1)
Diabetes Metab 2013. 39(2):89-93
|
|

|
The continuing need for drug development and clinical trials in type 2 diabetes and its complications: introduction to the RDS Special Issue
Raz I, Gallwitz B
Rev Diabet Stud 2011. 8(3):288-292
|
|
|


)
)
)

