Review
| Rev Diabet Stud,
2011,
8(3):355-368 |
DOI 10.1900/RDS.2011.8.355 |
Peptides and Their Potential Role in the Treatment of Diabetes and Obesity
Hannah C. Greenwood, Stephen R. Bloom, Kevin G. Murphy
Section of Investigative Medicine, Department of Medicine, Imperial College London, London, UK
Address correspondence to: Kevin Murphy, e-mail: k.g.murphy@imperial.ac.uk
Manuscript submitted June 30, 2011; resubmitted August 23, 2011; accepted September 3, 2011.
Keywords: gastrointestinal hormone, hypothalamus, neuropeptide, obesity, type 2 diabetes
Abstract
It is estimated that 347 million people worldwide have diabetes and that over 1.5 billion adults worldwide are overweight. Predictions suggest these rates are increasing. Diabetes is a common complication in overweight and obese subjects, and in 2004, an estimated 3.4 million people died from consequences of high blood sugar. Thus, there is great interest in revealing the physiological systems that regulate body weight and blood sugar. Several peptidergic systems within the central nervous system and the periphery regulate energy homeostasis. A number of these systems have been investigated as potential treatments for obesity and the metabolic syndrome. However, manipulation of peptidergic systems poses many problems. This review discusses the peptidergic systems currently attracting research interest for their clinical potential to treat obesity. We consider first neuropeptides in the brain, including the orexigenic neuropeptide Y and melanin-concentrating hormone, and anorectic factors such as the melanocortins, ciliary neurotrophic factor, and neuromedin U. We subsequently discuss the utility of targeting peripheral gut peptides, including pancreatic polypeptide, peptide YY, amylin, and the gastric hormone ghrelin. Also, we analyze the evidence that these factors or drugs based on them may be therapeutically useful, while considering the disadvantages of using such peptides in a clinical context.
Abbreviations: α-MSH - alpha-melanocyte-stimulating hormone; ACTH - adrenocorticotropic hormone; AgRP - agouti-related peptide; AMRI - Albany Molecular Research Inc.; ARC - arcuate nucleus; BMS - Bristol-Myers Squibb; C57BL/6 - inbred strain C57 black 6; CNS - central nervous system; CNTF - ciliary neurotrophic factor; DIO - diet-induced obesity; GHS-R - growth hormone secretagogue receptor; GI - gastrointestinal; GLP-1 - glucagon-like peptide 1; ICV - intracerebroventricular; MC1R - melanocortin 1 receptor; MCH - melanin-concentrating hormone; MCH1R - MCH 1 receptor ; mRNA - messenger ribonucleic acid; MSH - melanocyte-stimulating hormone; MTII - melanotan II; NDP - 4Nle-D-7Phe; NMU - neuromedin U; NMU-R1 - neuromedin U receptor 1; NPY - neuropeptide Y; PEG - polyethylene glycol; POMC - pro-opiomelanocortin; PP - pancreatic polypeptide; PYY - peptide YY; SNAP- 7941 - selective, non-peptide antagonist at the MCH receptor; STAT3 - signal transducer and activator of transcription 3
Introduction
The metabolic syndrome is a collection of medical disorders that together contribute to an increased risk of developing diabetes and heart disease. It is estimated that 347 million people worldwide have diabetes, and in 2004, an estimated 3.4 million people died from consequences of high blood sugar [1]. The metabolic syndrome has many pseudonyms and definitions, and the underlying cause is not completely understood. Although the metabolic syndrome is comprised of many factors, most, if not all, can be improved with weight loss. Over 1.5 billion adults worldwide are overweight. Predictions suggest these rates are increasing. Excess weight and diabetes are conditions associated with a high morbidity and mortality [2].
Understanding the regulation of body weight is thus vital to identify potential therapeutic targets. The control of body weight involves a coordinated regulation of both food intake and energy expenditure to promote stable levels of energy storage in organs such as adipose tissue and liver. This process, referred to as energy homeostasis, is the result of integrating neural and hormonal signals from the periphery and neural signals within the central nervous system (CNS) to defend a relatively consistent body weight. The efficiency of this system is demonstrated by the fact that, following a period of fasting, food intake increases until body weight recovers, and it returns to baseline levels [3]. The effects of incretins on the regulation of energy homeostasis are extensively discussed in other articles within this issue. The purpose of this article is to discuss other peptidergic systems involved in the regulation of energy homeostasis and their therapeutic potential.
Central peptides regulating energy homeostasis
Energy homeostasis is regulated by peptides synthesized and released within the CNS, which act as neurotransmitters, and by peptide hormones produced peripherally, for example by the gastrointestinal (GI) tract, which act either via the vagus nerve or directly on the CNS (see Figure 1). Within the CNS, the hypothalamus is a key region in the regulation of energy homeostasis. The brain stem also plays a role; it receives several signals:
1. Neural signals from the gut via the vagus nerve.
2. Hormonal signals.
Subsequently, the brain stem signals to the hypothalamus to influence appetite.
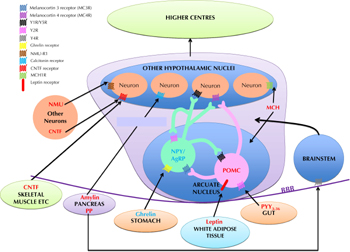 |
 |
Figure 1. Schematic diagram illustrating the peptidergic signaling pathways regulating energy homeostasis. Peptides colored blue are orexigenic, promoting hunger, whilst peptides colored red are anorexigenic, promoting satiety. POMC: pro-opiomelanocortin. NPY: neuropeptide Y. AgRP: agouti-related peptide. NMU: neuromedin U. CNTF: ciliary neurotrophic factor. MCH: melanin concentrating hormone. PYY3-36: peptide YY3-36. PP: pancreatic polypeptide. BBB: blood-brain barrier. |
|
The hypothalamus comprises several discrete nuclei. They are responsible for integrating and processing both central and peripheral signals of energy homeostasis, and for inducing the changes in feeding behavior and/or energy expenditure required to maintain overall energy homeostasis [4]. The hypothalamic nuclei communicate with each other and with other parts of the brain via release of specific neuropeptides. Some of these hypothalamic neuropeptide systems are putative targets for anti-obesity therapies. The potential systems and therapies are discussed in this article (see Table 1).
Table
1.
Drugs targeting peptidergic systems recently investigated for their potential as anti-obesity therapies |
|
 |
|
Legend:
All agents are believed to act in the central nervous system, in particular on the hypothalamus. * There are no published data regarding administration in humans; so animal data is given. |
|
Neuropeptide Y
Neuropeptide Y (NPY) is a 36 amino acid peptide. It is produced in several hypothalamic nuclei and is important in controlling energy homeostasis. The NPY-containing neuronal population in the hypothalamic arcuate nucleus (ARC) is the best characterized. The ARC is located adjacent to the median eminence, which has an incomplete blood brain barrier, facilitating sensing of peripheral nutrients and hormones. NPY is expressed in the medial ARC where it is colocalized with another orexigenic neuropeptide, agouti-related peptide (AgRP) [5, 6]. ARC neurones expressing NPY project to several other nuclei, including the paraventricular nucleus, dorsomedial nucleus, and lateral hypothalamic area [7, 8]. They also express receptors for peripheral signals of energy homeostasis, e.g. ghrelin and leptin [9-11].
NPY is a member of the PP-fold peptide family that includes the gut hormones peptide YY (PYY) and pancreatic polypeptide (PP), which are discussed below. The members of the PP-fold family signal through the Y receptors, Y1-Y5. NPY is thought to physiologically increase food intake. Hypothalamic NPY expression is upregulated by fasting, and central administration of NPY increases food intake by acting at the Y1 and Y5 receptors [4]. Although NPY, Y1, and Y5 knockout models do not have the profound alterations in energy homeostasis which might be expected, this is thought to be due to developmental compensation [12-14]. Ablation of ARC NPY/AgRP neurones in adult mice results in hypophagia and weight loss [15-17].
Y1 and Y5 receptor antagonists have thus been designed to antagonize the effects of endogenous NPY on appetite and food intake. Several Y1 receptor antagonists have been administered to rodents, but most have associated safety concerns, and none have been tested in humans [18, 19]. The Y1 receptor antagonist J-115814 inhibits feeding in lean C57BL/6 mice and in hyperphagic db/db mice, which lack leptin signaling, and in mice with diet-induced obesity (DIO) [20, 21]. However, J-115814 potently interacts with the IKr potassium channel [18], suggesting that it could have cardiovascular side effects. Another Y1 receptor antagonist, BMS-193885, attenuates both NPY-induced and spontaneous nocturnal feeding following intraperitoneal administration to rats [19]. Chronic administration of BMS-193885 suppressed feeding and body weight gain. However, although no behavioral side effects were identified, a number of rats fell ill following chronic administration [19]. Also, the drug is not orally available and induced adhesive peritonitis in these rats following intraperitoneal administration due to poor tissue absorption [19].
Several Y5 receptor antagonists have been demonstrated to be effective at reducing DIO, whilst others have shown no effect, as reviewed in [22]. Many of those which are effective have unwanted side effects [22]. Nonetheless, some Y5 receptor antagonists have been tested in humans. The orally active Y5 receptor antagonist MK-0557 induced weight loss statistically greater than placebo control after one year of treatment, but the magnitude of this effect was not clinically meaningful [23]. The orally active Y5 receptor antagonist S-2367 resulted in a small reduction in body weight compared to placebo-treated controls when administered to obese subjects in conjunction with a 500-800 kcal/day energy deficit [22]. S-2367 may be more effective at reducing body weight in conditions where NPY signaling might be expected to be higher, for example in ob/ob mice and in caloric restriction [22]. S-2367 initially appeared to be well-tolerated in humans. However, some studies reported a small, statistically insignificant, reduction in erythrocyte count, hemoglobin and hematocrit, raising concerns of a potential risk of anemia [22].
Therefore, it remains unclear whether antagonizing endogenous NPY receptor signaling will be useful for the treatment of obesity. NPY is the most abundant neuropeptide in the CNS. It is difficult to specifically target its hyperphagic action because of its wide range of functions. Therefore, it remains problematic to produce an agent that targets NPY signaling to reduce food intake without causing side effects.
Melanocortinergic signaling
The hypothalamic melanocortin system has a key role in the regulation of energy homeostasis [24]. Melanocortins are peptides produced from the gene encoding pro-opiomelanocortin (POMC). They exert their biological effects via five melanocortin receptors (MC1R-MC5R), both in the periphery and the CNS [25]. POMC mRNA levels are decreased in food-deprived rats [26]. Intracerebroventricular (ICV) administration of alpha-melanocyte-stimulating hormone (α-MSH), a 13 amino acid peptide product of POMC which acts as a ligand for the MC3R and MC4R, reduces food intake in rats [27-29]. The physiological importance of melanocortin signaling in body weight maintenance has been demonstrated using a number of animal models; knockout of POMC or MC4R results in obesity [30, 31]. It is thought that α-MSH provides a physiological anorectic tone at the MC4R that restrains food intake. Deletion of MC3R does not result in obesity, but does influence energy homeostasis [32, 33].
Melanotan II (MTII), a melanocortin receptor agonist originally developed as a tanning agent, reduces food intake when injected peripherally to rodents, suggesting it can cross the blood brain barrier [34]. The effects of MTII on food intake appear to be mediated via the MC4R; MTII has approximately a five-fold higher potency at the mouse MC4R versus the MC3R, and MTII does not have anorectic effects in MC4R null mice [35]. Most studies that pharmacologically manipulate central melanocortin signaling to reduce food intake have been conducted in rodents. There is only one published study in non-human primates which shows an anorectic effect of ICV NDP-MSH, a stable α-MSH analogue [36].
The melanocortin system is also important in humans. Individuals with mutations in POMC are hyperphagic and obese, and haploinsufficiency of MC4R has been shown to be the most common known monogenic cause of human obesity [37-39]. Other genetic mutations in melanocortinergic pathways have been linked with obesity [40, 41]. Despite this convincing evidence that MC4R is critical in energy homeostasis in humans, it has not been demonstrated that administration of an MC4R agonist can treat human obesity. Intranasal administration of ACTH/MSH4-10 (representing the core sequence of all melanocortins) reduces food intake in normal weight males, but is ineffective in obese males, and chronic treatment does not reduce body weight in children with POMC deficiency [42-44]. It is therefore possible that obesity induces resistance to the effects of MC4R agonists, worsening obesity and invalidating MC4R agonism as a potential therapy. However, it must be noted that ACTH/MSH4-10 lacks potent activity at MC4R [45]. In a 12 week study of obese human subjects, potent agonism of MC4R with MK-0493 did not reduce energy intake or body weight compared to placebo [46]. This is further evidence that melanocortin resistance may occur, although the compound was not tested in lean individuals. Also, MC4R agonism has been shown to induce off target effects in humans, including detrimental effects in blood pressure and sympathetic function [47]. Further research is required to determine whether such side effects limit the use of MC4R agonists for the treatment of obesity, or whether it is possible to selectively target MC4R appetite pathways.
Melanin-concentrating hormone signaling
Melanin-concentrating hormone (MCH) was originally isolated from the pituitary gland of teleost fish, in which it controls skin pigmentation [48]. MCH was subsequently discovered to be expressed in the hypothalamus and zona incerta [49]. MCH is overexpressed in the hypothalamus of rodents following fasting and in obese ob/ob mice [50]. Central administration of MCH increases food intake in rats [50]. Also, mice which lack the precursor to MCH are hypophagic and lean [51]. The MCH receptor MCH1R is widely expressed in central and peripheral tissues. The expression pattern of MCH1R suggests that MCH is involved in many physiological processes [52, 53]. Mice with targeted deletion of MCH1R are hyperphagic [54], implicating this receptor in the regulation of energy homeostasis. A further MCH receptor, MCH2R, has been identified in humans, but is not expressed in rodents [55, 56].
In 2002, two papers reported that small molecule antagonists of MCH1R might be useful in the treatment of obesity [57, 58]. Acute intracerebroventricular administration of SNAP-7941 or T-226296 decreased MCH-induced food intake in rats, and chronic administration of SNAP-7941 reduced body weight in rats with DIO [57, 58]. Several MCH1R antagonists have since been created and shown to be effective at reducing body weight in animal models of DIO, as reviewed in [59].
These effects may not be exclusively mediated through the CNS. Peripheral MCH signaling modulates leptin secretion and insulin release [60, 61]. Systemic MCH1R antagonism may thus modulate energy homeostasis via both central and peripheral mechanisms. MCH1R antagonist-induced weight loss is suggested to be driven by increased energy expenditure in addition to reduced food intake. Administration of an MCH1R antagonist to mice moderately suppressed feeding but resulted in a disproportionately large reduction in body weight [62]. Pair feeding to the MCH1R-treated group resulted in a smaller reduction in body weight, suggesting that MCH1R antagonism also has effects on energy expenditure [62].
One MCH1R antagonist, ALB-127158(a), has recently completed Phase I trials in humans, with promising results. Reports from the pharmaceutical company AMRI have stated that ALB-127158(a) dose-dependently reduced food intake in DIO mice in preclinical testing. The resultant weight loss of up to 18% after 28 days of administration was substantially higher than that achieved with sibutramine. Weight loss correlated with a reduction in food intake, a preferential reduction in fat stores, and improvements in glucose tolerance. At the doses tested in humans, ALB-127158(a) was well tolerated, and subjects reported reduced appetite, hunger, and desire to eat. Subjects who received the compound consumed less at a test meal [63]. Further trials will identify whether ALB-127158(a) can reduce food intake in obese subjects, and whether it promotes weight loss when administered chronically.
Ciliary neurotrophic factor
Ciliary neurotrophic factor (CNTF) was initially described as having trophic actions on motor neurones in the ciliary ganglion, and to influence other motor neurone populations [64, 65]. Clinical trials were conducted to determine whether CNTF is beneficial in patients suffering from motor neurone disease in the mid 1990's. An unexpected outcome was that CNTF induced substantial weight loss. This finding was initially treated with caution, as it was suspected that CNTF might be acting in a manner similar to cachectic cytokines such as interleukin-1. However, subsequent studies suggested that CNTF may play a physiological role in food intake. CNTF and leptin signaling pathways share molecular components, including the STAT3 transcription factor [66, 67]. CNTF receptors are present in hypothalamic nuclei important in feeding, and CNTF causes preferential loss of fat in ob/ob mice [68]. Importantly, CNTF is also effective in DIO, a state which is similar to human obesity, and which results in resistance to the anorectic effects of leptin [68].
A phase III trial for the use of axokine, a modified version of human CNTF, for the treatment of obesity was initiated. Although there was a small improvement in some patients, approximately 70% of subjects developed antibodies against the drug after three months of treatment [69]. Those who did not develop antibodies lost around 12.5 pounds in a year compared to 4.5 pounds in the placebo group. However, it was feared that development of antibodies to a CNTF-derived molecule might reduce the neuroprotective effects of endogenous CNTF [69], and axokine was abandoned as an anti-obesity agent.
Neuromedin U
Neuromedin U (NMU) is a member of the neuromedin peptide family. It was originally isolated from porcine spinal cord and subtyped ‘U’ because of its potent ability to cause uterine contraction [70]. NMU is widely expressed throughout the body, with highest expression in the brain and GI tract [71]. NMU does not circulate, and is thought to act as a neuropeptide rather than as a hormone. Hypothalamic NMU mRNA levels are significantly reduced following fasting [72]. ICV administration of NMU reduces food intake in mice [73] and rats [72]. NMU administration also increases both physical activity and non-exercise activity thermogenesis in both DIO and lean rats [74]. NMU null mice display hyperphagia, increased body weight and adiposity, and decreased locomotor activity and energy expenditure, suggesting a role for this peptide in the regulation of energy homeostasis [75]. These mice also develop late-onset hyperglycemia and hyperlipidemia [75]. Transgenic mice that overexpress NMU are hypophagic, and have reduced body weight, low somatic and liver fat, and improved insulin sensitivity [76]. On a high fat diet, these mice remain lighter than wild types and eat less [76]. A variant in the human NMU gene has also been linked to obesity in the human population [77].
NMU signals via two receptors, NMU-R1 and NMU-R2. The distribution of these receptors has not been clearly defined. However, NMU-R1 is thought to be expressed predominantly in the periphery, particularly in the GI tract, while NMU-R2 is expressed in hypothalamic nuclei known to regulate energy homeostasis [72, 78]. NMU reduces food intake when administered acutely or chronically, and these effects have been determined to require NMU-R1 signaling. The development of NMU-R1 agonists may therefore prove effective in the treatment of obesity [79]. As there is evidence that NMU increases energy expenditure and improves glucose tolerance, such agents might also have positive effects on other aspects of the metabolic syndrome [79]. However, NMU is implicated in the regulation of other physiological systems, including stress, and it is possible that the use of NMU-R1 antagonists may thus have side effects [80].
Peripheral peptides regulating energy homeostasis
A number of peripherally produced peptide hormones signal to the CNS both directly and indirectly, via the vagus nerve, to influence energy homeostasis. The GI tract is a major site of the production of hormones which influence energy homeostasis. Hormones produced in the gut such as glucagon-like peptide 1 (GLP-1) and oxyntomodulin, which reduce food intake and improve glycemic control, are discussed in other articles in this issue. However, the gut synthesizes and releases a number of other hormones that affect food intake. Peptide or protein hormones which regulate appetite are also produced in additional peripheral sites around the body, in particular the endocrine pancreas. These hormone systems and their potential as anti-obesity targets are discussed below.
PP-fold gut peptides
The PP-fold peptide family includes NPY and the gut hormones PYY and PP. The orexigenic effects of NPY are mediated by central Y1 and Y5 receptors, as discussed above. In contrast, the Y2 and Y4 receptors mediate anorectic effects of PYY and PP, respectively [81-83]. PYY is an anorectic gut hormone expressed in enteroendocrine L-cells throughout the GI tract, with highest tissue content in the terminal ileum, colon, and rectum [84, 85]. PYY exists in two forms. The amino terminal of the full-length peptide PYY1-36 is cleaved by the enzyme dipeptidyl peptidase IV (DPP-IV) to form the amino-terminally truncated form PYY3-36, which comprises the majority of PYY circulating immunoreactivity [86, 87]. PYY1-36 binds with relatively high affinity to all five Y receptors (Y1-Y5), whereas PYY3-36 is a selective Y2 receptor agonist [82].
PYY is released into the circulation following a meal at levels proportional to calories consumed [84], and circulating levels are reduced by fasting. Peripheral administration of PYY3-36 reduces food intake in wild-type rodents and lean and obese humans, but not in mice lacking the Y2 receptor [81, 88]. PP is synthesized and released postprandially from the endocrine pancreas in proportion to the calorie content of a meal, and is postulated to act as a satiety signal via activation of the Y4 receptor. Peripheral administration of PP to both rodents and humans also reduces food intake [89, 90].
While studies demonstrate that Y2 and Y4 agonists can reduce appetite acutely, the circulating half-lives of PYY and PP are short, suggesting that the native peptides may be difficult to use as agents to chronically reduce body weight. Also, it has been shown that central administration of PYY3-36 or PP in rodents stimulates appetite [91-94]. This is thought to be due to high local concentrations of these hormones pharmacologically stimulating orexigenic Y receptors. This suggests that sufficiently high peripheral doses of PYY and PP might result in orexigenic effects. In addition, the therapeutic window for PYY3-36 is relatively narrow, with low doses proving ineffective and high doses causing nausea [95].
Preprandial intranasal PYY3-36 was tested in obese patients for two weeks, but was not found to reduce body weight. Intranasal administration resulted in transiently high circulating levels of PYY associated with nausea [96]. PYY analogues selective for the Y2 receptor, and which show moderately increased but persistent circulating levels following administration, may be more useful in the treatment of obesity. A PEGylated selective Y2 receptor agonist has been shown to outperform PYY3-36 in reducing food intake in mice [97], suggesting that PEGylation of specific Y2 receptor agonists may improve their therapeutic potential.
PP has not been found to cause nausea so it is possible that PP will prove a more useful therapeutic target than PYY [90]. Also, PP-releasing neuroendocrine tumors are not associated with nausea [98]. A fatty acid acylated human PP analogue specific for Y2 and Y4 receptors had improved pharmacokinetic properties compared to a non-lipidated human PP analogue [99]. The lipidation also increased the duration of food intake reduction in mice, suggesting that it might be useful in the treatment of obesity [99]. Thus, structural modification and acylation have proved useful as tools in increasing the efficacy of PP-fold gut peptides. The Y4 receptor agonist TM30339 has proved efficacious at reducing body weight in diet-induced obese mice, and is currently undergoing phase I/II trials [100]. The same pharmaceutical company is also carrying out phase I/II trials of a dual Y2 and Y4 receptor agonist, obinepitide, which has been shown to inhibit food intake in obese individuals up to nine hours after subcutaneous administration [101].
Amylin
Amylin, or islet amyloid polypeptide, is a peptide hormone secreted by pancreatic β-cells concurrently with insulin to synergistically control blood glucose levels [102]. It activates the calcitonin receptor when dimerized with a receptor activity-modulating protein [103]. In addition to its effects on blood glucose, peripheral administration of amylin reduces food intake in wild type, ob/ob, and db/db mice [104]. Rats receiving daily doses of amylin for 6 days prior to the dark phase had a reduced food intake and body weight [105]. Amylin also slows gastric emptying, and there is evidence that it enhances the action of other peptides such as cholecystokinin.
The clinical utility of amylin is restricted by its instability and its tendency to self-aggregate. Many diabetics are deficient in amylin. Pramlintide (symlin), a synthetic analogue of amylin which is soluble and non-aggregating, is currently approved to be used with insulin by type 1 and type 2 diabetics. Treatment of type 1 and type 2 diabetic patients with symlin at doses which mimic the plasma levels of non-diabetics for one year results in sustained weight loss [106, 107]. Symlin is currently being tested in non-diabetics as a treatment for obesity. One disadvantage of using symlin as an anti-obesity drug is that it has to be injected at meal times, rather than using more user-friendly administration protocols.
A recent study has suggested that amylin might be used in combination with leptin as a weight-loss therapy. Leptin is a protein adipostat hormone produced in adipose tissue which circulates at levels proportional to body fat to regulate energy homeostasis [108, 109]. Concurrent infusion of amylin and leptin to DIO rats reduces food intake and body weight, specifically fat mass [110, 111]. Similar results have also been demonstrated in humans; coadministration of symlin and the leptin analogue metreleptin to overweight/obese subjects results in a greater weight loss than either analogue alone [110]. It has been suggested that amylin might restore or enhance sensitivity to leptin in obesity [112, 113]. However, this development program has now been abandoned at phase II. It has been shown in rats that removal of either amylin or leptin results in regain of all body weight lost following amylin and leptin coadministration, indicating that both agents are required [114]. It is possible that using other anorectic gut hormones in combination with leptin might also demonstrate additive or synergistic effects. Combination therapies are an attractive strategy, as weight loss evokes powerful compensatory mechanisms which are responsible for recovering the lost body weight [115]. Targeting multiple signaling systems may attenuate the effects of these compensatory pathways. However, current drug-testing legislation does not facilitate the identification of novel effective drug combinations for the treatment of obesity, or indeed for any disease.
Ghrelin
Ghrelin is synthesized in the stomach and is the only gastrointestinal hormone known to stimulate appetite [116]. It was first discovered as the endogenous ligand for the growth hormone secretagogue receptor (GHS-R) [117]. Ghrelin is also expressed in the hypothalamic ARC and periventricular area [117, 118]. The GHS-R is expressed in many tissues throughout the body, including the gut, hypothalamus, and pituitary [119]. Ghrelin levels rise before meals and fall postprandially [120]. Acutely, central or peripheral administration of ghrelin potently stimulates feeding in rodents [121]. Chronic administration induces weight gain [116]. Intravenous ghrelin infusion increases energy consumption during a buffet meal in humans [122].
Ghrelin acts via the orexigenic ARC NPY neurones in the hypothalamus [123, 124], though it may also act on the vagal brainstem pathway in addition to having a direct effect [123]. It is also thought to target the mesolimbic dopamine system to enhance food reward. Ghrelin and GHS-R knockout mice are resistant to DIO. Ghrelin also inhibits glucose-stimulated insulin release. Therefore, antagonizing ghrelin signaling could improve glucose tolerance [125]. Blocking GHS-R signaling has thus been proposed as a treatment for obesity and diabetes.
Ghrelin spiegelmers (single stranded oligonucleotide 'mirror images' of the peptide to be inhibited) have been created to inhibit the function of endogenous ghrelin. Intravenous administration to rats of NOX-B11, a ghrelin spiegelmer, suppressed ghrelin-induced neuronal activity and ghrelin-induced food intake [126]. Another novel approach to reduce the effect of circulating ghrelin is the use of anti-ghrelin vaccines. Rats injected with rat ghrelin linked to a large immunogenic carrier molecule mounted an immune response to ghrelin. After one week, rats with higher ghrelin-specific antibody titres gained less weight and had lower levels of body fat [127]. However, such vaccination is theoretically permanent and so there is a danger that too much body weight could be lost, with detrimental effects on health. A number of GHS-R antagonists have also been described, but have not been demonstrated to reduce food intake in animals.
Ghrelin is implicated in other physiological systems besides energy homeostasis, including growth, learning, and blood glucose regulation [117, 123, 125], which may result in side effects for agents modulating the ghrelin system. Although the animal evidence supporting the use of agents blocking GHS-R signaling is compelling, the efficacy of ghrelin antagonists has yet to be proven in clinical trials. Ghrelin levels are reduced in obese subjects [128], which may explain why it is difficult to reduce obesity using this strategy. It is possible that such agents may be useful in conditions of hyperghrelinemia such as Prader-Willi syndrome.
Conclusions
A number of peptidergic systems have been shown to influence food intake and energy homeostasis. Manipulation of several of these systems reduces food intake and body weight in preclinical models. However, in clinical trials, many of these agents failed to meaningfully reduce body weight [46, 69, 96]. This is due to a number of reasons, including lack of efficacy and side effects. These two issues are related; it may be impossible to administer such an agent at the dose required to reduce body weight because of the associated side effects. The relatively wide expression of many peptidergic system components, and their associated diversity of function, makes their manipulation for the treatment of obesity fraught with difficulties.
There are also difficulties with the routes of administration. For example, agents targeting CNS receptors must have a means of crossing the blood brain barrier. If a drug needs to be taken often, the route of administration becomes a patient issue. It is inconvenient and painful to have to inject a drug; orally active small molecule treatments are preferable. However, by their very nature, peptide hormones are difficult to mimic with small molecules. In particular, many gut hormone receptors require large ligands for receptor interaction, and residues essential for receptor interaction can be widely distributed throughout their secondary structure.
Furthermore, antibodies may develop against drugs based on peptides. This carries the risk that the immune response extends to the endogenous peptide, as shown for axokine. As most of these peptidergic systems have multiple functions, this could have extremely deleterious effects. Nevertheless, despite these difficulties, recent studies have produced promising results. Therefore, peptidergic systems remain an attractive research target in the fight against obesity.
Disclosures: The authors report no conflict of interests.
Acknowledgments:
The Section is funded by grants from the MRC, BBSRC, NIHR, an Integrative Mammalian Biology (IMB) Capacity Building Award, an FP7- HEALTH-2009-241592 EuroCHIP grant, and is supported by the NIHR Imperial Biomedical Research Centre Funding Scheme. Kevin G. Murphy is supported by project grants from the NIHR and BBSRC.
References
- Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011. 378(9785):31-40. [DOD] [CrossRef]
- WHO. Obesity and overweight. 2011. [DOD]
- Harris RB, Kasser TR, Martin RJ. Dynamics of recovery of body composition after overfeeding, food restriction or starvation of mature female rats. J Nutr 1986. 116(12):2536-2546. [DOD]
- Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev 1999. 20(1):68-100. [DOD] [CrossRef]
- Broberger C, Landry M, Wong H, Walsh JN, Hokfelt T. Subtypes Y1 and Y2 of the neuropeptide Y receptor are respectively expressed in pro-opiomelanocortin- and neuropeptide-Y-containing neurons of the rat hypothalamic arcuate nucleus. Neuroendocrinology 1997. 66(6):393-408. [DOD] [CrossRef]
- Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci 1998. 1(4):271-272. [DOD] [CrossRef]
- Chronwall BM, DiMaggio DA, Massari VJ, Pickel VM, Ruggiero DA, O'Donohue TL. The anatomy of neuropeptide-Y-containing neurons in rat brain. Neuroscience 1985. 15(4):1159-1181. [DOD] [CrossRef]
- Morris BJ. Neuronal localisation of neuropeptide Y gene expression in rat brain. J Comp Neurol 1989. 290(3):358-368. [DOD] [CrossRef]
- Willesen MG, Kristensen P, Romer J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 1999. 70(5):306-316. [DOD] [CrossRef]
- Baskin DG, Schwartz MW, Seeley RJ, Woods SC, Porte D Jr, Breininger JF, Jonak Z, Schaefer J, Krouse M, Burghardt C, et al. Leptin receptor long-form splice-variant protein expression in neuron cell bodies of the brain and co-localization with neuropeptide Y mRNA in the arcuate nucleus. J Histochem Cytochem 1999. 47(3):353-362. [DOD] [CrossRef]
- Baskin DG, Breininger JF, Schwartz MW. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes 1999. 48(4):828-833. [DOD] [CrossRef]
- Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, et al. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol 2002. 22(14):5027-5035. [DOD] [CrossRef]
- Pedrazzini T, Seydoux J, Kunstner P, Aubert JF, Grouzmann E, Beermann F, Brunner HR. Cardiovascular response, feeding behavior and locomotor activity in mice lacking the NPY Y1 receptor. Nat Med 1998. 4(6):722-726. [DOD] [CrossRef]
- Marsh DJ, Hollopeter G, Kafer KE, Palmiter RD. Role of the Y5 neuropeptide Y receptor in feeding and obesity. Nat Med 1998. 4(6):718-721. [DOD] [CrossRef]
- Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, Ghatei MA, Bloom SR. Post-embryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J 2005. 19(12): 1680-1682. [DOD]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci 2005. 8(10):1289-1291. [DOD] [CrossRef]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 2005. 310(5748):683-685. [DOD] [CrossRef]
- Kameda M, Kobayashi K, Ito H, Miyazoe H, Tsujino T, Nakama C, Kawamoto H, Ando M, Ito S, Suzuki T, et al. Optimization of a series of 2,4-diaminopyridines as neuropeptide Y Y1 receptor antagonists with reduced hERG activity. Bioorg Med Chem Lett 2009. 19(15):4325-4329. [DOD] [CrossRef]
- Antal-Zimanyi I, Bruce MA, Leboulluec KL, Iben LG, Mattson GK, McGovern RT, Hogan JB, Leahy CL, Flowers SC, Stanley JA, et al. Pharmacological characterization and appetite suppressive properties of BMS-193885, a novel and selective neuropeptide Y(1) receptor antagonist. Eur J Pharmacol 2008. 590(1-3):224-232. [DOD] [CrossRef]
- Kanatani A, Hata M, Mashiko S, Ishihara A, Okamoto O, Haga Y, Ohe T, Kanno T, Murai N, Ishii Y, et al. A typical Y1 receptor regulates feeding behaviors: effects of a potent and selective Y1 antagonist, J-115814. Mol Pharmacol 2001. 59(3):501-505. [DOD]
- Mashiko S, Moriya R, Ishihara A, Gomori A, Matsushita H, Egashira S, Iwaasa H, Takahashi T, Haga Y, Fukami T, et al. Synergistic interaction between neuropeptide Y1 and Y5 receptor pathways in regulation of energy homeostasis. Eur J Pharmacol 2009. 615(1-3):113-117. [DOD] [CrossRef]
- Yulyaningsih E, Zhang L, Herzog H, Sainsbury A. NPY receptors as potential targets for anti-obesity drug development. Br J Pharmacol 2011. 163(6):1170-1202. [DOD] [CrossRef]
- Erondu N, Gantz I, Musser B, Suryawanshi S, Mallick M, Addy C, Cote J, Bray G, Fujioka K, Bays H, et al. Neuropeptide Y5 receptor antagonism does not induce clinically meaningful weight loss in overweight and obese adults. Cell Metab 2006. 4(4):275-282. [DOD] [CrossRef]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci 2005. 8(5):571-578. [DOD] [CrossRef]
- Cone RD, Lu D, Koppula S, Vage DI, Klungland H, Boston B, Chen W, Orth DN, Pouton C, Kesterson RA. The melanocortin receptors: agonists, antagonists, and the hormonal control of pigmentation. Recent Prog Horm Res 1996. 51:287-317. [DOD]
- Brady LS, Smith MA, Gold PW, Herkenham M. Altered expression of hypothalamic neuropeptide mRNAs in food-restricted and food-deprived rats. Neuroendocrinology 1990. 52(5):441-447. [DOD] [CrossRef]
- Poggioli R, Vergoni AV, Bertolini A. ACTH-(1-24) and alpha-MSH antagonize feeding behavior stimulated by kappa opiate agonists. Peptides 1986. 7(5):843-848. [DOD] [CrossRef]
- Tsujii S, Bray GA. Acetylation alters the feeding response to MSH and beta-endorphin. Brain Res Bull 1989. 23(3):165-169. [DOD] [CrossRef]
- Brown KS, Gentry RM, Rowland NE. Central injection in rats of alpha-melanocyte-stimulating hormone analog: effects on food intake and brain Fos. Regul Pept 1998. 78(1-3):89-94. [DOD] [CrossRef]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997. 88(1):131-141. [DOD] [CrossRef]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 1999. 5(9):1066-1070. [DOD] [CrossRef]
- Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 2000. 141(9):3518-3521. [DOD] [CrossRef]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet 2000. 26(1):97-102. [DOD] [CrossRef]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 1997. 385(6612):165-168. [DOD] [CrossRef]
- Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, Marsh DJ, Forrest MJ, Gopal-Truter S, Fisher J, et al. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res 2000. 9(2):145-154. [DOD] [CrossRef]
- Koegler FH, Grove KL, Schiffmacher A, Smith MS, Cameron JL. Central melanocortin receptors mediate changes in food intake in the rhesus macaque. Endocrinology 2001. 142(6):2586-2592. [DOD] [CrossRef]
- Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, O'Rahilly S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest 2000. 106(2):271-279. [DOD] [CrossRef]
- Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 2000. 106(2):253-262. [DOD] [CrossRef]
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998. 19(2):155-157. [DOD] [CrossRef]
- Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O'Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet 1997. 16(3):303-306. [DOD] [CrossRef]
- Lee YS, Poh LK, Kek BL, Loke KY. The role of melanocortin 3 receptor gene in childhood obesity. Diabetes 2007. 56(10):2622-2630. [DOD] [CrossRef]
- Fehm HL, Smolnik R, Kern W, McGregor GP, Bickel U, Born J. The melanocortin melanocyte-stimulating hormone/adrenocorticotropin(4-10) decreases body fat in humans. J Clin Endocrinol Metab 2001. 86(3):1144-1148. [DOD] [CrossRef]
- Hallschmid M, Smolnik R, McGregor G, Born J, Fehm HL. Overweight humans are resistant to the weight-reducing effects of melanocortin4-10. J Clin Endocrinol Metab 2006. 91(2):522-525. [DOD] [CrossRef]
- Krude H, Biebermann H, Schnabel D, Tansek MZ, Theunissen P, Mullis PE, Gruters A. Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4-10. J Clin Endocrinol Metab 2003. 88(10):4633-4640. [DOD] [CrossRef]
- Schioth HB, Muceniece R, Wikberg JE. Characterisation of the melanocortin 4 receptor by radioligand binding. Pharmacol Toxicol 1996. 79(3):161-165. [DOD] [CrossRef]
- Krishna R, Gumbiner B, Stevens C, Musser B, Mallick M, Suryawanshi S, Maganti L, Zhu H, Han TH, Scherer L, et al. Potent and selective agonism of the melanocortin receptor 4 with MK-0493 does not induce weight loss in obese human subjects: energy intake predicts lack of weight loss efficacy. Clin Pharmacol Ther 2009. 86(6):659-666. [DOD] [CrossRef]
- Greenfield JR. Melanocortin signalling and the regulation of blood pressure in human obesity. J Neuroendocrinol 2011. 23(2):186-193. [DOD] [CrossRef]
- Kawauchi H, Kawazoe I, Tsubokawa M, Kishida M, Baker BI. Characterization of melanin-concentrating hormone in chum salmon pituitaries. Nature 1983. 305(5932):321-323. [DOD] [CrossRef]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol 1992. 319(2):218-245. [DOD] [CrossRef]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 1996. 380(6571):243-247. [DOD] [CrossRef]
- Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 1998. 396(6712):670-674. [DOD] [CrossRef]
- Saito Y, Nothacker HP, Wang Z, Lin SH, Leslie F, Civelli O. Molecular characterization of the melanin-concentrating-hormone receptor. Nature 1999. 400(6741):265-269. [DOD] [CrossRef]
- Saito Y, Cheng M, Leslie FM, Civelli O. Expression of the melanin-concentrating hormone (MCH) receptor mRNA in the rat brain. J Comp Neurol 2001. 435(1):26-40. [DOD] [CrossRef]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A 2002. 99(5):3240-3245. [DOD] [CrossRef]
- Hill J, Duckworth M, Murdock P, Rennie G, Sabido-David C, Ames RS, Szekeres P, Wilson S, Bergsma DJ, Gloger IS, et al. Molecular cloning and functional characterization of MCH2, a novel human MCH receptor. J Biol Chem 2001. 276(23):20125-20129. [DOD] [CrossRef]
- Sailer AW, Sano H, Zeng Z, McDonald TP, Pan J, Pong SS, Feighner SD, Tan CP, Fukami T, Iwaasa H, et al. Identification and characterization of a second melanin-concentrating hormone receptor, MCH-2R. Proc Natl Acad Sci U S A 2001. 98(13):7564-7569. [DOD] [CrossRef]
- Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, et al. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med 2002. 8(8):825-830. [DOD]
- Takekawa S, Asami A, Ishihara Y, Terauchi J, Kato K, Shimomura Y, Mori M, Murakoshi H, Suzuki N, Nishimura O, et al. T-226296: a novel, orally active and selective melanin-concentrating hormone receptor antagonist. Eur J Pharmacol 2002. 438(3):129-135. [DOD] [CrossRef]
- Luthin DR. Anti-obesity effects of small molecule melanin-concentrating hormone receptor 1 (MCHR1) antagonists. Life Sci 2007. 81(6):423-440. [DOD] [CrossRef]
- Bradley RL, Kokkotou EG, Maratos-Flier E, Cheatham B. Melanin-concentrating hormone regulates leptin synthesis and secretion in rat adipocytes. Diabetes 2000. 49(7):1073-1077. [DOD] [CrossRef]
- Tadayyon M, Welters HJ, Haynes AC, Cluderay JE, Hervieu G. Expression of melanin-concentrating hormone receptors in insulin-producing cells: MCH stimulates insulin release in RINm5F and CRI-G1 cell-lines. Biochem Biophys Res Commun 2000. 275(2):709-712. [DOD] [CrossRef]
- Ito M, Ishihara A, Gomori A, Matsushita H, Metzger JM, Marsh DJ, Haga Y, Iwaasa H, Tokita S, Takenaga N, et al. Mechanism of the anti-obesity effects induced by a novel melanin-concentrating hormone 1-receptor antagonist in mice. Br J Pharmacol 2010. 159(2):374-283. [DOD] [CrossRef]
- AMRI, phase 1 data support the continued development of ALB-127158(a) as a potential treatment for obesity. 2011. [DOD]
- Lin LF, Armes LG, Sommer A, Smith DJ, Collins F. Isolation and characterization of ciliary neurotrophic factor from rabbit sciatic nerves. J Biol Chem 1990. 265(15):8942-8947. [DOD]
- Sendtner M, Arakawa Y, Stockli KA, Kreutzberg GW, Thoenen H. Effect of ciliary neurotrophic factor (CNTF) on motoneuron survival. J Cell Sci Suppl 1991. 15:103-109. [DOD]
- Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995. 267(5202):1349-1353. [DOD] [CrossRef]
- Lambert PD, Anderson KD, Sleeman MW, Wong V, Tan J, Hijarunguru A, Corcoran TL, Murray JD, Thabet KE, Yancopoulos GD, et al. Ciliary neurotrophic factor activates leptin-like pathways and reduces body fat, without cachexia or rebound weight gain, even in leptin-resistant obesity. Proc Natl Acad Sci U S A 2001. 98(8):4652-4657. [DOD] [CrossRef]
- Gloaguen I, Costa P, Demartis A, Lazzaro D, Di Marco A, Graziani R, Paonessa G, Chen F, Rosenblum CI, Van der Ploeg LH, et al. Ciliary neurotrophic factor corrects obesity and diabetes associated with leptin deficiency and resistance. Proc Natl Acad Sci U S A 1997. 94(12):6456-6461. [DOD] [CrossRef]
- Ettinger MP, Littlejohn TW, Schwartz SL, Weiss SR, McIlwain HH, Heymsfield SB, Bray GA, Roberts WG, Heyman ER, Stambler N, et al. Recombinant variant of ciliary neurotrophic factor for weight loss in obese adults: a randomized, dose-ranging study. JAMA 2003. 289(14):1826-1832. [DOD] [CrossRef]
- Minamino N, Kangawa K, Matsuo H. Neuromedin U-8 and U-25: novel uterus stimulating and hypertensive peptides identified in porcine spinal cord. Biochem Biophys Res Commun 1985. 130(3):1078-1085. [DOD] [CrossRef]
- Domin J, Ghatei MA, Chohan P, Bloom SR. Neuromedin U - a study of its distribution in the rat. Peptides 1987. 8(5):779-784. [DOD] [CrossRef]
- Howard AD, Wang R, Pong SS, Mellin TN, Strack A, Guan XM, Zeng Z, Williams DL Jr, Feighner SD, Nunes CN, et al. Identification of receptors for neuromedin U and its role in feeding. Nature 2000. 406(6791):70-74. [DOD] [CrossRef]
- Hanada T, Date Y, Shimbara T, Sakihara S, Murakami N, Hayashi Y, Kanai Y, Suda T, Kangawa K, Nakazato M. Central actions of neuromedin U via corticotropin-releasing hormone. Biochem Biophys Res Commun 2003. 311(4):954-958. [DOD] [CrossRef]
- Novak CM, Zhang M, Levine JA. Sensitivity of the hypothalamic paraventricular nucleus to the locomotor-activating effects of neuromedin U in obesity. Brain Res 2007. 1169:57-68. [DOD] [CrossRef]
- Hanada R, Teranishi H, Pearson JT, Kurokawa M, Hosoda H, Fukushima N, Fukue Y, Serino R, Fujihara H, Ueta Y, et al. Neuromedin U has a novel anorexigenic effect independent of the leptin signaling pathway. Nat Med 2004. 10(10):1067-1073. [DOD] [CrossRef]
- Kowalski TJ, Spar BD, Markowitz L, Maguire M, Golovko A, Yang S, Farley C, Cook JA, Tetzloff G, Hoos L, et al. Transgenic overexpression of neuromedin U promotes leanness and hypophagia in mice. J Endocrinol 2005. 185(1):151-164. [DOD] [CrossRef]
- Hainerova I, Torekov SS, Ek J, Finkova M, Borch-Johnsen K, Jorgensen T, Madsen OD, Lebl J, Hansen T, Pedersen O. Association between neuromedin U gene variants and overweight and obesity. J Clin Endocrinol Metab 2006. 91(12):5057-5063. [DOD] [CrossRef]
- Wren AM, Small CJ, Abbott CR, Jethwa PH, Kennedy AR, Murphy KG, Stanley SA, Zollner AN, Ghatei MA, Bloom SR. Hypothalamic actions of neuromedin U. Endocrinology 2002. 143(11):4227-4234. [DOD] [CrossRef]
- Peier AM, Desai K, Hubert J, Du X, Yang L, Qian Y, Kosinski JR, Metzger JM, Pocai A, Nawrocki AR, et al. Effects of Peripherally Administered Neuromedin U on Energy and Glucose Homeostasis. Endocrinology 2011. In press. [DOD]
- Budhiraja S, Chugh A. Neuromedin U: physiology, pharmacology and therapeutic potential. Fundam Clin Pharmacol 2009. 23(2):149-157. [DOD] [CrossRef]
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature 2002. 418(6898):650-654. [DOD] [CrossRef]
- Abbott CR, Small CJ, Kennedy AR, Neary NM, Sajedi A, Ghatei MA, Bloom SR. Blockade of the neuropeptide Y Y2 receptor with the specific antagonist BIIE0246 attenuates the effect of endogenous and exogenous peptide YY(3-36) on food intake. Brain Res 2005. 1043(1-2):139-144. [DOD] [CrossRef]
- Lin S, Shi YC, Yulyaningsih E, Aljanova A, Zhang L, Macia L, Nguyen AD, Lin EJ, During MJ, Herzog H, et al. Critical role of arcuate Y4 receptors and the melanocortin system in pancreatic polypeptide-induced reduction in food intake in mice. PLoS One 2009. 4(12):e8488. [DOD] [CrossRef]
- Adrian TE, Ferri GL, Bacarese-Hamilton AJ, Fuessl HS, Polak JM, Bloom SR. Human distribution and release of a putative new gut hormone, peptide YY. Gastroenterology 1985. 89(5):1070-1077. [DOD]
- Ekblad E, Sundler F. Distribution of pancreatic polypeptide and peptide YY. Peptides 2002. 23(2):251-261. [DOD] [CrossRef]
- Grandt D, Schimiczek M, Beglinger C, Layer P, Goebell H, Eysselein VE, Reeve JR Jr. Two molecular forms of peptide YY (PYY) are abundant in human blood: characterization of a radioimmunoassay recognizing PYY 1-36 and PYY 3-36. Regul Pept 1994. 51(2):151-159. [DOD] [CrossRef]
- Medeiros MD, Turner AJ. Processing and metabolism of peptide-YY: pivotal roles of dipeptidylpeptidase-IV, aminopeptidase-P, and endopeptidase-24.11. Endocrinology 1994. 134(5):2088-2094. [DOD] [CrossRef]
- Batterham RL, Cohen MA, Ellis SM, Le Roux CW, Withers DJ, Frost GS, Ghatei MA, Bloom SR. Inhibition of food intake in obese subjects by peptide YY3-36. N Engl J Med 2003. 349(10):941-948. [DOD] [CrossRef]
- Asakawa A, Inui A, Yuzuriha H, Ueno N, Katsuura G, Fujimiya M, Fujino MA, Niijima A, Meguid MM, Kasuga M. Characterization of the effects of pancreatic polypeptide in the regulation of energy balance. Gastroenterology 2003. 124(5):1325-1336. [DOD] [CrossRef]
- Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M, Frost GS, Ghatei MA, Bloom SR. Pancreatic polypeptide reduces appetite and food intake in humans. J Clin Endocrinol Metab 2003. 88(8):3989-3992. [DOD] [CrossRef]
- Clark JT, Sahu A, Kalra PS, Balasubramaniam A, Kalra SP. Neuropeptide Y (NPY)-induced feeding behavior in female rats: comparison with human NPY ((Met17)NPY), NPY analog ((norLeu4)NPY) and peptide YY. Regul Pept 1987. 17(1):31-39. [DOD] [CrossRef]
- Corp ES, McQuade J, Krasnicki S, Conze DB. Feeding after fourth ventricular administration of neuropeptide Y receptor agonists in rats. Peptides 2001. 22(3):493-499. [DOD] [CrossRef]
- Clark JT, Kalra PS, Crowley WR, Kalra SP. Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 1984. 115(1):427-429. [DOD] [CrossRef]
- Asakawa A, Inui A, Ueno N, Fujimiya M, Fujino MA, Kasuga M. Mouse pancreatic polypeptide modulates food intake, while not influencing anxiety in mice. Peptides 1999. 20(12):1445-1448. [DOD] [CrossRef]
- Degen L, Oesch S, Casanova M, Graf S, Ketterer S, Drewe J, Beglinger C. Effect of peptide YY3-36 on food intake in humans. Gastroenterology 2005. 129(5):1430-1436. [DOD] [CrossRef]
- Gantz I, Erondu N, Mallick M, Musser B, Krishna R, Tanaka WK, Snyder K, Stevens C, Stroh MA, Zhu H, et al. Efficacy and safety of intranasal peptide YY3-36 for weight reduction in obese adults. J Clin Endocrinol Metab 2007. 92(5):1754-1757. [DOD] [CrossRef]
- DeCarr LB, Buckholz TM, Milardo LF, Mays MR, Ortiz A, Lumb KJ. A long-acting selective neuropeptide Y2 receptor PEGylated peptide agonist reduces food intake in mice. Bioorg Med Chem Lett 2007. 17(7):1916-1919. [DOD] [CrossRef]
- Adrian TE, Uttenthal LO, Williams SJ, Bloom SR. Secretion of pancreatic polypeptide in patients with pancreatic endocrine tumors. N Engl J Med 1986. 315(5):287-291. [DOD] [CrossRef]
- Bellmann-Sickert K, Elling CE, Madsen AN, Little PB, Lundgren K, Gerlach LO, Bergmann R, Holst B, Schwartz TW, Beck-Sickinger AG. Long-acting lipidated analogue of human pancreatic polypeptide is slowly released into circulation. J Med Chem 2011. 54(8):2658-2667. [DOD] [CrossRef]
- Pharma T. TM30339. 2011. [DOD]
- Pharma T. Obinepitide. 2011. [DOD]
- Pittner RA, Albrandt K, Beaumont K, Gaeta LS, Koda JE, Moore CX, Rittenhouse J, Rink TJ. Molecular physiology of amylin. J Cell Biochem 1994. 55(Suppl):19-28. [DOD] [CrossRef]
- Sexton PM, Albiston A, Morfis M, Tilakaratne N. Receptor activity modifying proteins. Cell Signal 2001. 13(2):73-83. [DOD] [CrossRef]
- Morley JE, Flood JF, Horowitz M, Morley PM, Walter MJ. Modulation of food intake by peripherally administered amylin. Am J Physiol 1994. 267(1 Pt 2):R178-R184. [DOD]
- Morris MJ, Nguyen T. Does neuropeptide Y contribute to the anorectic action of amylin? Peptides 2001. 22(3):541-546. [DOD]
- Whitehouse F, Kruger DF, Fineman M, Shen L, Ruggles JA, Maggs DG, Weyer C, Kolterman OG. A randomized study and open-label extension evaluating the long-term efficacy of pramlintide as an adjunct to insulin therapy in type 1 diabetes. Diabetes Care 2002. 25(4):724-730. [DOD] [CrossRef]
- Ratner R, Whitehouse F, Fineman MS, Strobel S, Shen L, Maggs DG, Kolterman OG, Weyer C. Adjunctive therapy with pramlintide lowers HbA1c without concomitant weight gain and increased risk of severe hypoglycemia in patients with type 1 diabetes approaching glycemic targets. Exp Clin Endocrinol Diabetes 2005. 113(4):199-204. [DOD] [CrossRef]
- Badman MK, Flier JS. The adipocyte as an active participant in energy balance and metabolism. Gastroenterology 2007. 132(6):2103-2115. [DOD] [CrossRef]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996. 334(5):292-295. [DOD] [CrossRef]
- Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, Anderson CM, Parkes DG, Baron AD. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A 2008. 105(20):7257-7262. [DOD] [CrossRef]
- Trevaskis JL, Coffey T, Cole R, Lei C, Wittmer C, Walsh B, Weyer C, Koda J, Baron AD, Parkes DG, et al. Amylin-mediated restoration of leptin responsiveness in diet-induced obesity: magnitude and mechanisms. Endocrinology 2008. 149(11):5679-5687. [DOD] [CrossRef]
- Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am J Physiol Regul Integr Comp Physiol 2004. 286(1):R143-R150. [DOD] [CrossRef]
- Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience 2001. 104(4):1111-1117. [DOD] [CrossRef]
- Trevaskis JL, Lei C, Koda JE, Weyer C, Parkes DG, Roth JD. Interaction of leptin and amylin in the long-term maintenance of weight loss in diet-induced obese rats. Obesity (Silver Spring) 2010. 18(1):21-26. [DOD] [CrossRef]
- Chen HC, Roth JD, Schroeder BE, Weyer C. Role of islet-, gut-, and adipocyte-derived hormones in the central control of food intake and body weight: implications for an integrated neurohormonal approach to obesity pharmacotherapy. Curr Diabetes Rev 2008. 4(2):79-91. [DOD] [CrossRef]
- Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature 2000. 407(6806):908-913. [DOD] [CrossRef]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999. 402(6762):656-660. [DOD] [CrossRef]
- Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003. 37(4):649-661. [DOD] [CrossRef]
- Hosoda H, Kojima M, Kangawa K. Biological, physiological, and pharmacological aspects of ghrelin. J Pharmacol Sci 2006. 100(5):398-410. [DOD] [CrossRef]
- Cummings DE, Weigle DS, Frayo RS, Breen PA, Ma MK, Dellinger EP, Purnell JQ. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 2002. 346(21):1623-1630. [DOD] [CrossRef]
- Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA, et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 2000. 141(11):4325-4328. [DOD] [CrossRef]
- Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 2001. 86(12):5992. [DOD] [CrossRef]
- Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S. A role for ghrelin in the central regulation of feeding. Nature 2001. 409(6817):194-198. [DOD] [CrossRef]
- Wang L, Saint-Pierre DH, Tache Y. Peripheral ghrelin selectively increases Fos expression in neuropeptide Y - synthesizing neurons in mouse hypothalamic arcuate nucleus. Neurosci Lett 2002. 325(1):47-51. [DOD] [CrossRef]
- Tong J, Prigeon RL, Davis HW, Bidlingmaier M, Kahn SE, Cummings DE, Tschop MH, D'Alessio D. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 2010. 59(9):2145-2151. [DOD] [CrossRef]
- Kobelt P, Helmling S, Stengel A, Wlotzka B, Andresen V, Klapp BF, Wiedenmann B, Klussmann S, Monnikes H. Anti-ghrelin Spiegelmer NOX-B11 inhibits neurostimulatory and orexigenic effects of peripheral ghrelin in rats. Gut 2006. 55(6):788-792. [DOD] [CrossRef]
- Zorrilla EP, Iwasaki S, Moss JA, Chang J, Otsuji J, Inoue K, Meijler MM, Janda KD. Vaccination against weight gain. Proc Natl Acad Sci U S A 2006. 103(35):13226-13231. [DOD] [CrossRef]
- Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes 2001. 50(4):707-709. [DOD] [CrossRef]
This article has been cited by other articles:

|
Position and length of fatty acids strongly affect receptor selectivity pattern of human pancreatic polypeptide analogues
Mäde V, Bellmann-Sickert K, Kaiser A, Meiler J, Beck-Sickinger AG
ChemMedChem 2014. 9(11):2463-2474
|
|
|
Immuno-modulator metallo-peptide reduces inflammatory state in obese zucker fa/fa rats
Gomez-Solis A, Reyes-Esparza J, Garcia-Vazquez F, Alvarez-Ayala E, Rodriguez-Fragoso L
Int J Biomed Sci 2014. 10(3):172-181
|
|
|
The SCFA Receptor GPR43 and Energy Metabolism
Kimura I, Inoue D, Hirano K, Tsujimoto G
Front Endocrinol (Lausanne) 2014. 5:85
|
|

|
Amylin under examination. Fibrillogenic polypeptide hormone of the pancreas
Marszalek M
Postepy Hig Med Dosw 2014. 68:29-41
|
|

|
Recombinant production of peptide C-terminal α-amides using an engineered intein
Albertsen L, Shaw AC, Norrild JC, Stromgaard K
Bioconjug Chem 2013. 24(11):1883-1894
|
|

|
The modulatory role of high fat feeding on gastrointestinal signals in obesity
Duca FA, Sakar Y, Covasa M
J Nutr Biochem 2013. 24(10):1663-1677
|
|

|
Pathomechanisms of the development of obesity in some endocrinopathies - an overview
Pujanek M, Bronisz A, Małecki P, Junik R
Endokrynol Pol 2013. 64(2):150-155
|
|

|
Outstanding scientific achievement award lecture 2011: defeating diabesity: the case for personalized combinatorial therapies
Tschöp MH, DiMarchi RD
Diabetes 2012. 61(6):1309-1314
|
|

|
The continuing need for drug development and clinical trials in type 2 diabetes and its complications: introduction to the RDS Special Issue
Raz I, Gallwitz B
Rev Diabet Stud 2011. 8(3):288-292
|
|
|


)
)

