Review
| Rev Diabet Stud,
2011,
8(4):454-467 |
DOI 10.1900/RDS.2011.8.454 |
Hepatic Steatosis in Type 1 Diabetes
Simon E. Regnell, Ake Lernmark
Lund University, CRC, Department of Clinical Sciences, Diabetes and Celiac Disease Unit, Skane University Hospital, SUS SE-20502, Malmö, Sweden
Address correspondence to: Simon E. Regnell, e-mail: fek09sre@student.lu.se
Manuscript submitted January 7, 2012; resubmitted January 20, 2012; accepted February 5, 2012.
Keywords: type 1 diabetes, hepatic steatosis, non-alcoholic fatty liver disease, NAFLD, hepatic fat, glycogen synthesis, insulin, hepatocyte
Abstract
Islet autoimmunity in type 1 diabetes results in the loss of the pancreatic β-cells. The consequences of insulin deficiency in the portal vein for liver fat are poorly understood. Under normal conditions, the portal vein provides 75% of the liver blood supply. Recent studies suggest that non-alcoholic fatty liver disease (NAFLD) may be more common in type 1 diabetes than previously thought, and may serve as an independent risk marker for some chronic diabetic complications. The pathogenesis of NAFLD remains obscure, but it has been hypothesized that hepatic fat accumulation in type 1 diabetes may be due to lipoprotein abnormalities, hyperglycemia-induced activation of the transcription factors carbohydrate response element-binding protein (ChREBP) and sterol regulatory element-binding protein 1c (SREBP-1c), upregulation of glucose transporter 2 (GLUT2) with subsequent intrahepatic fat synthesis, or a combination of these mechanisms. Novel approaches to non-invasive determinations of liver fat may clarify the consequences for liver metabolism when the pancreas has ceased producing insulin. This article aims to review the factors potentially contributing to hepatic steatosis in type 1 diabetes, and to assess the feasibility of using liver fat as a prognostic and/or diagnostic marker for the disease. It provides a background and a case for possible future studies in the field.
Abbreviations: ACC - acetyl-CoA carboxylase; ALAT - alanine transaminase; ALP - alkaline phosphatase; ASAT - aspartate aminotransferase; ATP - adenosine triphosphate; BMI - body mass index; ChREBP - carbohydrate response element-binding protein; CoA - coenzyme A; FAS - fatty acid synthase; FATP - fatty acid transport protein; GFR - glomerular filtration rate; GGT - γ-glutamyltransferase; GLUT2 - glucose transporter 2; HbA1c - glycated hemoglobin; HDL - high-density lipoprotein; IDE - insulin-degrading enzyme; IRS-1/2 - insulin receptor substrate 1/2; LDL - low-density lipoprotein; LPK - liver-type pyruvate kinase; LXRα - liver X receptor α; MAPK - mitogen-activating protein kinases; MRI - magnetic resonance imaging; mRNA - messenger ribonucleic acid; NAFLD - non-alcoholic fatty liver disease; PI3K - phosphatidylinositol-3 kinase; PP - pancreatic polypeptide; Shc - Src homologous and collagen; SREBP-1c - sterol regulatory element-binding protein 1c; UDP - uridine diphosphate; VLDL - very low-density lipoprotein
Introduction
Accumulation of liver fat in patients with type 1 diabetes has long been reported [1]. However, in contrast to the attention given to non-alcoholic fatty liver disease in type 2 diabetes [2, 3], the etiology, prevalence, and consequences of hepatic steatosis in type 1 diabetes remain poorly understood. The lack of attention could be attributed to the greater prevalence of type 2 diabetes as compared to type 1 diabetes, and the association of type 2 diabetes with obesity, which is a well-established risk factor for fatty liver.
This article is organized as follows: the first section deals with physiological processes related to the hepatic handling of insulin which could be affected in type 1 diabetes and which play a role in non-alcoholic fatty liver disease (NAFLD). The second part investigates the molecular effects of insulin and hyperglycemia on hepatocytes to uncover enzymatic pathways that potentially contribute to NAFLD in type 1 diabetes. In the third section, evidence is provided on the prevalence of hepatic steatosis among type 1 diabetes patients. This is done by discussing the epidemiological studies carried on the subject, and compiling liver biopsy data from case reports. Furthermore, we discuss the correlation between NAFLD and diabetic complications, and we summarize the proposed biological mechanisms causing the condition. The fourth part of the article reviews hepatic carbohydrate metabolism in type 1 diabetes in order to expand on the theory that a derangement of this system could contribute to NAFLD. The fifth section comments on the effects of hormones other than insulin that are abnormal in type 1 diabetes to assess if any of them could potentially contribute to fatty liver. Finally, some of the possible clinical applications of further research on NAFLD in type 1 diabetes are discussed.
Handling of insulin in the liver
The passage of insulin to the liver
Blood that has passed through the pancreas enters the liver through the hepatic portal vein. The blood contains any insulin secreted by the β-cells of the pancreas. It is estimated that seventy-five percent of the dual blood flow to the liver is supplied by the hepatic portal vein, the remainder comes from the hepatic arteries [4]. The ratio of flow between the hepatic arteries and the portal vein is variable, and alters in response to stimuli such as food intake [5]. Changes in portal flow have been shown to affect the flow of the hepatic artery. When total portal flow is reduced, the flow in the hepatic artery increases rapidly. Conversely, an increase in portal flow leads to a reduction in arterial flow [6]. However, a change of the blood flow in the hepatic artery does not seem to affect the flow in the hepatic portal vein [7]. In rabbits, moderate steatosis reduced portal and total hepatic blood flow and microcirculation, with a significant increase in hepatic artery flow and portal pressure [8].
Despite the well-known fact that patients with long-standing type 1 diabetes no longer release insulin so that there is no insulin supply to the liver from the portal vein, there is a paucity of data on hepatic blood flow regulation in type 1 diabetes. The epidemiological prevalence and physiological effects of portal hypertension caused by type 1 diabetes-induced steatosis would merit investigation.
Dynamics of hepatic blood flow and insulin clearance
Blood from the portal vein mixes with blood from the hepatic artery in the sinusoids of the liver lobules. The sinusoids drain into the central vein of the lobule, and the central veins coalesce with the hepatic veins, which exit the liver. The fenestrated endothelium of the liver permits proteins, such as insulin, free access from the sinusoid into the space of Disse. Once proteins are inside the space of Disse, they can bind to cell surface receptors or be absorbed by the microvilli of the hepatocytes [9].
Roughly 50-80% of insulin that enters the liver through the portal vein is thought to be cleared during first-pass transit [10, 11]. The primary cellular mechanism for hepatic uptake and degradation of insulin is a receptor-mediated process. In addition to activating signal transduction pathways, the activated insulin receptor stimulates the internalization of itself and its bound hormone through endocytosis. The insulin molecule is released from the receptor by endosomes. It may then be degraded intracellularly, primarily by insulin-degrading enzyme, or released back into the circulation [12]. Finally, the receptors are recycled back to the plasma membrane [13].
Basically, insulin clearance in the liver is carried out by hepatocytes, with Kupffer cells only contributing to about 15% of the total hepatic insulin degradation. Non-receptor-mediated insulin uptake, called pinocytosis, may be significant in hepatocyte insulin uptake at high insulin concentrations [12].
The intracellular degradation of insulin is largely performed by insulin-degrading enzyme (IDE) [14]. IDE concentration generally correlates to the amount of insulin degradation, and the liver is the organ with the highest concentration of this enzyme [15]. In normal glucose concentrations, insulin has been shown to increase IDE activity in hepatocytes, thereby stimulating its own degradation [16].
Possible consequences of secretory oscillations and subcutaneous injection of insulin
The release of insulin from β-cells is biphasic. An increase in glucose results in an initial transient burst of insulin secretion that falls back to near-basal levels within ten minutes. The second phase involves a slower progression to maximal secretion levels, which persists throughout the duration of glucose exposure [17].
Both basal and postprandial circulating insulin levels oscillate with a period of approximately five to ten minutes [18], contemporaneously with the calcium concentration of the β-cells [19, 20]. The fluctuations are well synchronized entirely throughout the islets. This is because of the spread of electric potentials between adjacent cells through gap junctions [21, 22] and diffusible factors such as ATP [23]. In turn, the periodicities of the approximately one million islets in the human pancreas are thought to be synchronized by autonomic ganglia [24]. In addition to these aforementioned high-frequency fluctuations in insulin concentration, low-frequency fluctuations, with a period of 50 to 150 minutes, have also been reported in both humans and animals [25].
Both high-frequency and low-frequency fluctuations of insulin release may be biologically significant, since continuous insulin exposure induces downregulation of insulin receptor density on the target cell by internalization. The variations reduce this effect, thereby decreasing the amount of insulin needed to provide its cellular effects [26]. In experiments performed on rat livers, the potentiating effect of varying insulin levels was reflected by fluctuating insulin levels inhibiting gluconeogenesis more effectively than exposure to a constant concentration of the hormone [24].
Insulin replacement therapy in type 1 diabetes is based on subcutaneous administration of insulin [27]. Injected insulin is absorbed into the blood stream and a fraction of it reaches the liver both by the portal vein and the hepatic artery. It is unlikely that most of the injected insulin reaches the liver through the portal vein, as in normal conditions. It is more likely that the ratio between portal vein and hepatic artery insulin is closer to one. The consequence for blood flow independent of hepatocyte function in type 1 diabetes is not clear, but it cannot be excluded that liver fat and carbohydrate metabolism, as well as insulin degradation are affected.
Insulin action in the liver
Molecular effects of insulin
Regardless of the pathological setting, the normal effects of insulin are initiated by its binding to the transmembranous insulin receptor on the target cell. The insulin receptor consists of two polypeptide α-subunits, which are each linked to a β-subunit [28]. The extracellular α-subunits bind the insulin molecule [29]. Upon binding, they discontinue the inhibition of tyrosine kinase action of the transmembrane β-subunits [30].
The β-subunits phosphorylate tyrosines on the intracellular proteins insulin receptor substrate 1 and 2 (IRS-1 and IRS-2) and Src homologous and collagen (Shc) [31]. All of these three proteins eventually lead to the activation of mitogen-activating protein kinases (MAPK) [13], which in turn targets the cell nucleus, causing gene activation and transcription [32]. The IRS proteins also activate phosphatidylinositol-3 kinase (PI3K) [33], which has been linked to cell growth, proliferation, and survival [34]. is also assumed that PI3K causes translocation of glucose transporter type 4 (GLUT4) from internal vesicles to the cell membrane of fat and muscle cells [35], and protein synthesis [36].
Effects on carbohydrate metabolism
GLUT2 is a facilitative glucose transporter found primarily in the liver and pancreatic β-cells [37]. Several studies have demonstrated that liver GLUT2 expression is upregulated in a dose-dependent manner according to glucose concentration in primary rat hepatocytes and hepatocyte cell lines [38-40]. Also, liver GLUT2 expression in rats is reduced during starvation and increases to normal levels after refeeding [41]. In diabetic rats, liver GLUT2 expression is increased, but is corrected to regular levels when exogenous insulin returns the hyperglycemic conditions to normal [42]. These data suggest that GLUT2 expression in the liver is stimulated by glucose and downregulated by insulin. The possible implications of these actions are discussed below.
Furthermore, insulin decreases hepatic gluconeogenesis by suppressing gene expression of the key gluconeogenic enzymes phosphoenolpyruvate carboxykinase and glucose-6-phosphatase [43]. Also, insulin stimulates glycogen synthesis in the liver by activating glycogen synthetase [44]. Once the liver is saturated with glycogen, any additional glucose taken up by hepatocytes is moved to pathways leading to synthesis of fatty acids, which are exported from the liver as lipoproteins. In practice, however, saturation of glycogen stores requires a sustained overconsumption of carbohydrates for several days. Under normal conditions, the liver converts virtually no sugar to fat [45].
Stimulation of fat synthesis
Insulin has been shown to increase sterol regulatory element-binding proteins (SREBPs) in target cells, including hepatocytes. SREBPs are transcription factors that directly activate the expression of more than 30 genes dedicated to the synthesis and uptake of cholesterol, fatty acids, triglycerides, and phospholipids [46]. Insulin activates the aforementioned PI3K pathway, which leads to an augmented abundance of the precursor form of SREBP-1c in endoplasmic reticulum. This precursor form is then rapidly cleaved, leading to an increased content of the nuclear mature form of SREBP-1c [47]. A high glucose concentration has also been shown in vitro to upregulate SREBP-1c independently of insulin [48].
SREBP-1c is essential for glucokinase expression. In the presence of a high glucose level, and during the concerted action with the additional transcription factor carbohydrate response element-binding protein (ChREBP), SREBP-1c also assists the expression of the lipogenic genes liver-type pyruvate kinase (LPK), fatty acid synthase (FAS), and acetyl-CoA carboxylase (ACC) [49]. LPK catalyzes the conversion of phosphoenolpyruvate to pyruvate, the principal source of acetyl-CoA used for fatty acid synthesis [50]. FAS catalyzes the conversion of malonyl-CoA into long-chain saturated fatty acids [51]. ACC converts acetyl-CoA to malonyl-CoA. This in turn inhibits carnitine palmitoyl transferase-1-induced transport of fatty acids into mitochondria, thereby reducing mitochondrial fatty acid β-oxidation [52]. However, in primary hepatocytes, ChREBP can stimulate LPK gene transcription in response to high glucose concentrations without any apparent requirement for insulin [53]. ChREBP has been suggested as a contributor to fatty liver in the hyperglycemic setting of type 1 diabetes [54].
Insulin also induces expression of the transcription factor liver X receptor α (LXRα) in vivo [55]. One of two LXR isoforms, LXRα, is abundantly expressed in lipogenic tissues, and by activating the SREBP-1c promoter, it plays an important role in the transcriptional activation of lipogenic genes [56]. Another group of transcription factors in the liver, upstream stimulatory factors (USF), mediates the effects of insulin on the FAS promoter by interaction with SREBP-1c [57, 58].
In rodent hepatocytes, insulin affects steatosis by modulating free fatty acid flux via fatty acid transport proteins (FATPs) 2 and 5 in a U-shaped dose-dependent or bimodal fashion. This signaling is mediated via IRS-2 at low insulin concentrations, and through IRS-1at high insulin concentrations, indicating an optimal range of insulin levels. Deviations from this optimal range, whether hypoinsulinemia due to endogenous lack of insulin production or hyperinsulinemia due to excessive exogenous insulin administration, lead to increased hepatic triglyceride accumulation mediated by FATPs [59].
Liver fat in relation to a lack of insulin
Histology and prevalence of NAFLD
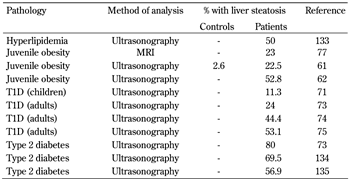
Nonalcoholic fatty liver disease (NAFLD) is a broad term, encompassing any net triglyceride retention in hepatocytes caused by any factor other than ethanol intake. NAFLD is histologically indistinguishable from the hepatic damage caused by alcohol abuse. Biopsy features include augmented triglyceride vacuoles, mixed inflammatory cell infiltration, hepatocyte ballooning and necrosis, glycogen nuclei, Mallory's hyaline, and fibrosis [60]. The condition is present in 10-24% of the general population in various countries [60]. It affects 2.6% of children in general [61], and between 22.5% [61] and 52.8% [62] of obese children. NAFLD has been reported in type 1 diabetes, but the mechanisms connecting the two conditions have not been investigated in detail. See Table 1 for a comparison of the prevalence of NAFLD in various pathologies.
Table
1.
Liver steatosis in conditions of hyperlipidemia, juvenile obesity, and diabetes |
|
 |
 |
Legend:
See references [61, 62, 71, 73-75, 77, 133-135] in reference list below. MRI: magnetic resonance imaging. T1D: type 1 diabetes. |
|
Increased liver fat has been associated with reduced insulin clearance and sensitivity [63]. It has also been linked to impaired insulin action to suppress hepatic glucose production in both non-diabetic subjects [64] and type 2 diabetic patients [65]. Insulin therapy has been shown to decrease liver fat content in type 2 diabetes patients [66], while in type 1 diabetes patients, liver fat correlates with poor glycemic control [67].
Liver biopsies from type 1 diabetes patients indicating hepatic steatosis
Despite advances in non-invasive medical imaging techniques, liver biopsy is still considered the most specific test to assess the severity and nature of liver disorders [68]. See Table 2 for a comparison of methods for liver fat analysis.
Table
2.
Methods of liver fat analysis |
|
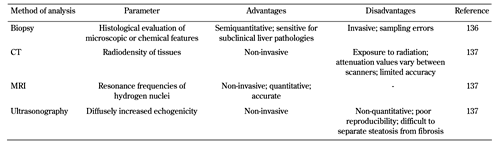 |
|
Legend:
See references [136] and [137] in reference list below. CT: computed tomography. MRI: magnetic resonance imaging. |
|
Generally, little information is available on liver biopsies from type 1 diabetes patients with NAFLD. Due to the absence of controlled studies on the subject, the only pertinent information we have found stems from six clinical case reports of patients presenting with severe diabetic symptoms due to poor glycemic control. These reports are summarized in Table 3.
Table
3.
Summary of case reports on liver biopsies from type 1 diabetes patients with NAFLD |
|
 |
|
Legend:
See references [54, 95, 103, 104, 138, 139] in reference list below. |
|
Prevalence of NAFLD in type 1 diabetes patients
Type 1 diabetes has been linked to NAFLD in children [69, 70], but only a few studies have aimed to determine the stability of the correlation between the two conditions. The prevalence of hepatic steatosis among type 1 diabetes patients was reported in five studies, all of which used ultrasonography to analyze the presence of liver fat. In a study of 106 children aged 8 months to 15 years, fatty liver was detected in 11.3% of the patients. The presence of fatty liver correlated to decreased glycemic control compared to patients without detectable fatty liver (mean HbA1c was 12.14% vs. 10.7%) [71]. Among 692 Egyptian type 1 diabetic children with a mean age of 9.65 ± 4.18 years, 4.5% had abnormal hyperechogenicity and/or hepatomegaly on ultrasound diagnostic [72].
In studies on adults with type 1 diabetes, the prevalence of NAFLD is markedly higher. A study of 17 patients with type 1 diabetes aged 65 ± 7 years detected hepatic steatosis in 24% of them, noting that patients with fatty livers had higher body mass indexes than their counterparts without steatosis [73]. Targher et al. observed fatty liver in 44.4% of a sample of 202 type 1 diabetes patients. Persons with NAFLD had higher HbA1c (8.4 ± 1.3 vs. 8.0 ± 1.1), lower glomerular filtration rate (GFR), longer duration of diabetes, and higher frequency of abnormal albuminuria, metabolic syndrome, and its individual components. Also, diabetes patients with NAFLD were older (aged 47 ± 12 years vs. 37 ± 12) and more likely to be male and obese than those without NAFLD [74]. In another sample of 343 type 1 diabetes patients, Targher et al. found a prevalence of 53.1% for hepatic steatosis. Again, persons with fatty liver had higher HbA1c (9.4 ± 2.2 vs. 9.1 ± 2.2), lower GFR, longer duration of diabetes, and higher frequency of abnormal albuminuria and the metabolic syndrome; were older (aged 39 ± 13 years vs. 49 ± 15), and more likely to be male and obese than those without NAFLD [75]. The differences in the prevalence of NAFLD between children and adults could be due to factors such as higher BMI [3] and increased duration of diabetes in adults.
A potential limitation in all five studies is that ultrasonography only can reliably ascertain a fat infiltration of more than 33% of hepatocytes [76]. Since non-obese healthy people have negligible amounts of liver fat (a fat fraction of over 9% has been called mild steatosis, while any value over 18% was considered severe steatosis [77]), the use of ultrasonography to determine the presence of NAFLD may not be reliable. It may fail to detect lower, but still pathologically elevated, hepatic fat levels, thus underestimating the prevalence of the condition. In any case, these studies contradict the notion that hepatic steatosis is rare in type 1 diabetes patients, occurring only in instances of extremely poor glycemic control.
Several epidemiological studies report a high incidence of the metabolic syndrome in type 1 diabetes patients (ranging from 20-50%) compared with non-diabetic subjects, which may provide indirect evidence of NAFLD [78-80]. Definitions of the metabolic syndrome vary, but a generally accepted characteristic of the state is insulin resistance [81]. Although hepatic insulin resistance is usually associated with type 2 diabetes, the condition also occurs in some type 1 diabetes patients [82]. Since insulin resistance has been associated with liver fat [83], this may provide an indication of the existence of NAFLD in type 1 diabetes patients. However, studies on the relationship between insulin resistance and hepatic steatosis in type 1 diabetes are lacking so far.
Elevated plasma levels of the liver enzyme alanine transaminase (ALAT) are an indicator of hepatocellular injury, and have been used as a marker for NAFLD [84], although other researchers have not found any clear correlation between NAFLD and abnormal ALAT concentrations [85, 86]. Two studies, relating serum ALAT content to type 1 diabetes, found elevated levels of the enzyme in 10-35% of type 1 diabetes patients, respectively [87, 88].
A study using ALAT, aspartate aminotransferase (ASAT), alkaline phosphatase (ALP), γ-glutamyltransferase (GGT), ferritin, and bilirubin as indicators of NAFLD found that ALAT, ASAT, ferritin, and the ASAT-to-ALAT ratio significantly correlated with insulin resistance in type 1 diabetes patients. In a logistic regression model, taking age, sex, BMI, and duration of diabetes into consideration, increased levels of ASAT, ALAT, and ALP were correlated with an increased risk of developing insulin resistance. The authors noted that the majority of their patients with the most severe insulin resistance had ultrasound-proven liver steatosis [89].
Over 50 gene loci have been found to influence the risk of type 1 diabetes [90]. There does not appear to be any research correlating any of the gene variants to liver fat content. This may be an interesting question to be clarified in future studies.
Relation of liver fat to diabetic complications
Type 1 diabetes is associated with a wide range of complications, including nephropathy, retinopathy, neuropathy, and macrovascular disease [91]. Since such long-term complications of type 1 diabetes develop gradually over years, the early detection of risk factors for and precursors of these conditions could facilitate their prognostication and treatment.
NAFLD has been correlated to cardiovascular disease in type 1 diabetes patients after adjustment for age, sex, smoking history, diabetes duration, HbA1c, LDL cholesterol, albuminuria, estimated glomerular filtration rate, and presence of the metabolic syndrome [74]. Because the study associated NAFLD with cardiovascular disease independently of these risk factors, it could be possible that NAFLD itself, at least in part, contributes to accelerated atherogenesis. The results, which demonstrated that NAFLD is associated with an increased prevalence of both asymptomatic and symptomatic cardiovascular disease in patients with type 1 diabetes, independently of several established risk factors, were later repeated with a larger sample of patients [92].
The same research group that conducted the aforementioned studies found that NAFLD was significantly associated with retinopathy and/or chronic kidney disease even after adjusting for age, sex, diabetes duration, HbA1c, body mass index, systolic blood pressure, triacylglycerol, medication use, and presence of the metabolic syndrome [93]. Again, due to the independence of NAFLD from these risk factors, it is possible that hepatic steatosis could be directly involved in the development of chronic kidney disease and retinopathy, although the possibility of reverse causality (i.e. kidney disease causing the development of NAFLD) cannot be definitely excluded. The team later produced similar results with a larger patient sample (343 compared to 202 in the earlier study), who attended a different clinic [75].
Abnormalities of lipoproteins in type 1 diabetes
In cases of well-controlled type 1 diabetes, plasma triglycerides and low-density lipoprotein (LDL) levels are normal or slightly decreased, while high density lipoproteins (HDL) are normal or slightly increased. However, in type 1 diabetes patients with poor glycemic control, plasma triglycerides and LDL levels are frequently increased [94].
Several potentially atherogenic qualitative abnormalities of lipoproteins are observed in patients with type 1 diabetes, even in those with good metabolic control. These abnormalities include increased cholesterol-to-triglyceride ratios in very low-density lipoprotein (VLDL), increased triglycerides in LDL and HDL, changes in the composition of the peripheral layer of lipoproteins, glycation of apolipoproteins, increased oxidation of LDL, and an increase in small dense LDL particles. These qualitative changes in lipoproteins are likely to impair their function. They cannot be fully explained by hyperglycemia and may be partly due to peripheral hyperinsulinemia associated with subcutaneous insulin administration [94]. It has been suggested that derangements of lipoproteins could be responsible for hepatic fat accumulation in type 1 diabetes [95]. Conversely, it is possible that NAFLD is not a consequence of, but rather a contributor to the development of lipid abnormalities.
Hypotheses for hepatic steatosis in type 1 diabetes
Based on the abovementioned findings, it seems that two hypotheses can be proposed to explain the occurrence of hepatic steatosis in type 1 diabetes in humans. First, atypical lipoprotein ratios and/or function could be responsible for an insufficient triglyceride secretion from the liver by VLDL. Second, transcription factors of hepatic metabolism such as ChREBP and SREBP-1c are activated by hyperglycemia and promote hepatic lipogenesis.
In addition, we may consider a third hypothesis, namely that hepatic steatosis may develop secondary to hyperglycemia in type 1 diabetes patients by glucose being transported from the blood into the liver by GLUT2, with the excess of sugar being converted into fat. This effect may be further amplified by hepatic upregulation of GLUT2 that occurs during hyperglycemia and hypoinsulinemia (characterizing precisely the conditions in type 1 diabetes), mediated by SREBP-1c [48], as described above. This hypothesis could be tested in part by determining the levels of GLUT2 protein and mRNA expression in hepatocytes from humans or animals with type 1 diabetes-induced fatty liver. See Figure 1 for a summary of the putative intracellular enzymatic mechanisms of NAFLD in type 1 diabetes.
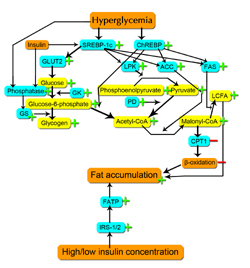 |
|
Figure 1. Proposed intracellular processes resulting in fatty liver in type 1 diabetes. Enzymes are in blue boxes, substrates in yellow, and miscellaneous processes in orange. Plus signs indicate increased activity; minus signs decreased activity. Abbreviations: SREBP-1c: sterol regulatory element-binding protein 1c. ChREBP: carbohydrate responsive element-binding protein. GLUT2: glucose transporter 2. LPK: liver-type pyruvate kinase. ACC: acetyl-CoA carboxylase. FAS: fatty acid synthase. GK: glucokinase. PD: pyruvate dehydroxygenase. GS: glycogen synthase. CPT1: carnitine palmitoyltransferase I. FATP: fatty acid transport protein. IRS: insulin receptor substrate. LCFA: long-chain fatty acid. |
|
Abnormal hepatic glycogen metabolism in type 1 diabetes
It may be regarded as possible that fat accumulation in the liver of type 1 diabetes patients is caused, in whole or in part, by conversion from carbohydrates. Then, one might expect to find abnormalities in the glycogen metabolism of the liver. Indeed, studies on this subject have found that patients with type 1 diabetes frequently have disturbances of hepatic glycogen accumulation and breakdown. However, the studies often appear contradictory.
Hepatic carbohydrate metabolism
A brief description of the main aspects of liver carbohydrate metabolism helps to evaluate the hypothesis that abnormal carbohydrate conversion causes fat accumulation in the liver. Hepatocytes take up glucose independently of insulin by the low-affinity, high-capacity glucose transporter GLUT2, which facilitates the entry of glucose in the presence of high concentrations of sinusoidal glucose. In the hepatocytes, glucose is rapidly phosphorylated to glucose-6-phosphate by the hepatic hexokinase isoform glucokinase. From glucose-6-phosphate, the glucose flux is directed into glycogen via uridine diphosphate (UDP)-glucose (direct pathway of glycogen synthesis), the pentose phosphate shunt, or into glycolysis, yielding molecules such as pyruvate and lactate [96]. Glycogen can also be synthesized by an indirect pathway (3 carbon units → phosphoenolpyruvate → glucose-6-phosphate → glucose-1-phosphate → UDP-glucose → glycogen) [97].
Glycogen synthesis and breakdown in type 1 diabetes
According to a study by Bischof et al., poorly controlled type 1 diabetic patients had a marked reduction in both hepatic glycogen synthesis and breakdown. Both defects in glycogen metabolism were improved, but not normalized, by short-term restoration of insulinemia and glycemia through intensified insulin treatment for 24 hours [98]. However, a subsequent study by the same researchers found that strict long- and short-term metabolic control using peripheral insulin substitution normalized hepatic glycogen synthesis, hepatic glycogenolysis, and nocturnal endogenous glucose production fasting in type 1 diabetic subjects. Yet, despite strenuous efforts to normalize glycemia, the contribution of the direct pathway to glycogen synthesis remained reduced in these patients, indicating augmented gluconeogenesis in type 1 diabetes, even after combined long- and short-term near normoglycemia. Hwang et al. performed a similar study on adults. They found that patients with poorly controlled type 1 diabetes stored only 30% as much glycogen during daytime as controls, with the indirect pathway of glycogen synthesis contributing to a proportionally greater part of glycogen synthesis, while their hepatic gluconeogenesis was increased [99]. The results were similar to previous findings from the same research group [97]. However, a study by Matyka et al. on prepubescent children found no such difference [100].
Inhibition of net hepatic glycogenolysis occurred in hypoglucagonic, hypoinsulinemic, and hyperglycemic conditions, mostly due to decreased glycogen phosphorylase flux [101]. In intensively treated type 1 diabetes patients, hypoglycemia failed to stimulate hepatic glycogen breakdown or activation of endogenous glucose production, despite activation of counterregulation. According to the authors, these factors may contribute to the defective hypoglycemic counterregulation seen in type 1 diabetes patients [102].
Cases of excessive glycogen storage in type 1 diabetes
Despite some previously mentioned studies showing that glycogen storage is decreased in persons with type 1 diabetes, there are numerous case reports of excessive glycogen storage in type 1 diabetes patients [103-112], some of the cases also had hepatic steatosis. The diabetes of all the patients that were examined due to palpable hepatomegaly was poorly controlled. However, this fact does not exclude that patients with a better control of their condition could have hepatic glycogen levels that are unusually high, but insufficient to cause hepatomegaly.
Possible mechanisms of hepatic glycogen accumulation in type 1 diabetes
It has been argued that hepatic glycogen accumulation in patients with unstable diabetes occurs due to an insulin-independent passive flux of glucose into the hepatocyte [104]. In the hepatocyte, glucose is converted irreversibly into glucose-6-phosphate by glucokinase, and subsequently trapped in the hepatocyte. Glucose-6-phosphate is then converted into glycogen by glycogen synthase. This enzyme exists in an inactive phosphorylated, and an active dephosphorylated form. The enzyme phosphatase is responsible for this conversion. Phosphatase concentration is maintained by insulin, and its activity relies on the presence of glucose. Thus, the synthesis of hepatic glycogen is promoted by high cytoplasmic glucose concentrations and relies on the presence of insulin. This is frequently seen in patients with unstable diabetes, where high blood glucose concentrations are associated with infrequent doses of insulin [104].
It has also been argued that patients promote hepatic glycogen accumulation when they take excess insulin and treat the subsequent hypoglycemic episode by administering glucose. Thus, a vicious cycle of hyperglycemia and intermittent supraphysiological insulin administration would result in glycogenosis [104, 113]. This theory could explain the observation by Nakamuta et al. that the vigorous treatment of diabetic ketoacidosis with insulin seemed to trigger liver enlargement [107]. We have not found any studies on the possible further conversion of hepatic carbohydrates to fat specifically in type 1 diabetes, but hepatocytes are generally capable of synthesizing fatty acids from glucose via acetyl-CoA, a reaction mediated by the aforementioned, hyperglycemia-activated transcription factor ChREPB [51].
Effects of other diabetes-associated endocrine disorders on the liver
In addition to losing their insulin production, type 1 diabetes patients may also suffer from additional disorders of pancreatic hormone secretion, affecting blood levels of glucagon, amylin, C-peptide, and pancreatic polypeptide.
Glucagon
Glucagon is secreted by the α-cells of the islets of Langerhans [114]. This secretion is suppressed independently by both hyperglycemia and the paracrine function of insulin [115], which may explain the relative excess of glucagon levels that occurs in diabetes patients [116]. Replacement with exogenous insulin to compensate the body’s own lack of insulin production in type 1 diabetes does not approach the paracrine levels of secreted insulin from β-cells, making this type of therapy relatively ineffective for lowering glucagon levels [117].
Glucagon increases hepatic gluconeogenesis and glucose output into the bloodstream, while reducing the liver’s glucose uptake and storage [118], inhibiting the incorporation of acetate into fatty acids and cholesterol [119], and causing hepatic lipolysis and ketogenesis [120]. These effects seem to counteract any hepatic anabolism. Indeed, exercise-stimulated glucagon receptor activation was found to be essential for treating high-fat diet-induced NAFLD in non-diabetic mice [121]. This further complicates the understanding of liver steatosis and glycogen accumulation in type 1 diabetes.
Amylin
Amylin is secreted concomitantly with insulin from the pancreatic β-cells. Therefore, individuals with type 1 diabetes are deficient in this hormone [122]. Among its effects in the immediate postprandial period, amylin may suppress glucagon secretion, reducing hepatic glucose production [123]. As the effects of amylin seem to be largely agonistic to those of insulin, a loss of the former hormone might possibly further aggravate the consequences of a deficit of the latter.
C-peptide
Insulin release from the pancreas may be measured by catheterizing the hepatic portal vein or measuring peripheral C-peptide concentrations. Since C-peptide is produced in equimolar amounts as insulin, yet not extracted by the liver, it provides an indicator for insulin secretion and hepatic clearance [124].
In addition to serving as a peripheral marker of insulin production, C-peptide may also have some biological functions in itself. For example, C-peptide was reported to stimulate Na+-K+-ATPase activity both in vitro and in vivo, elicit release of nitric oxide from endothelial cells, and mimic the effects of insulin in some tissues [125]. However, it is not clear whether C-peptide has any effect on liver function [126].
Pancreatic polypeptide
Pancreatic polypeptide (PP) is a hormone that arises from both the PP-cells of the islets of Langerhans and the acinar cells of the pancreas [127]. The secretion of this hormone is regulated by vagal cholinergic mechanisms in response to food intake and plasma glucose [128]. It has been shown that patients with type 1 diabetes release less pancreatic polypeptide after a meal than healthy persons, although their fasting levels of the hormone were similar [129]. Plasma PP has been shown to be clearly elevated in cases of diabetic ketoacidosis, and decreased following treatment with insulin and fluid infusion [130].
Binding proteins for PP have been identified on hepatocytic plasma membranes in rats [131]. PP administration in fed rats resulted in significantly increased hepatic insulin receptor concentration as compared to that seen in saline-administered fed animals [132]. The effects of PP on the liver in man seem to be poorly understood; relating the aforementioned studies to hepatic steatosis seems tenuous.
Future studies and clinical applications
Despite an abundance of studies on NAFLD, the prevalence, etiology, and consequences of the condition in conjunction with type 1 diabetes seem to have received relatively little attention. However, the few studies that have been done suggest that the state may be more common than generally assumed.
Studies linking liver fat to cardiovascular and kidney disease could potentially be advanced to provide a novel way of prognosticating complications of type 1 diabetes. Ultrasonography could be used to inexpensively screen patients for severe NAFLD, and to monitor the effects of treatment of liver steatosis by improving glycemic control. Further studies in the field may clarify the feasibility of using hepatic fat as a proxy for insulin production in the pancreas. If these two factors are sufficiently linked, liver fat measurements could potentially provide an alternative non-invasive method for prognosticating and diagnosing the presence and control of type 1 diabetes. For detailed definition, liver fat measurements could be carried out by magnetic resonance imaging (MRI) to provide a more precise determination of fat infiltration than ultrasonography.
Disclosure: The authors report no conflict of interests in relation to this work.
Acknowledgments:
The authors wish to thank Helena Elding Larsson and Pernilla Petersson for their feedback and advice.
References
- Connor CL. Fatty infiltration of the liver and the development of cirrhosis in diabetes and chronic alcoholism. Am J Pathol 1938. 14(3):347-364. [DOD]
- Roden M. Mechanisms of Disease: hepatic steatosis in type 2 diabetes - pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab 2006. 2(6):335-348. [DOD] [CrossRef]
- Smith BW, Adams LA. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat Rev Endocrinol 2011. 7(8):456-465. [DOD] [CrossRef]
- Greenway CV, Stark RD. Hepatic vascular bed. Physiol Rev 1971. 51:23-65. [DOD]
- Dauzat M, Lafortune M, Patriquin H, Pomier-Layrargues G. Meal induced changes in hepatic and splanchnic circulation: a noninvasive Doppler study in normal humans. Eur J Appl Physiol Occup Physiol 1994. 68(5):373-380. [DOD] [CrossRef]
- Rocheleau B, Ethier C, Houle R, Huet PM, Bilodeau M. Hepatic artery buffer response following left portal vein ligation: its role in liver tissue homeostasis. Am J Physiol 1999. 277(5 Pt 1):G1000-G1007. [DOD]
- Jakab F, Rath Z, Schmal F, Nagy P, Faller J. The interaction between hepatic arterial and portal venous blood flows; simultaneous measurement by transit time ultrasonic volume flowmetry. Hepatogastroenterology 1995. 42:18-21. [DOD]
- Seifalian AM, El-Desoky A, Davidson BR. Hepatic indocyanine green uptake and excretion in a rabbit model of steatosis. Eur Surg Res 2001. 33(3):193-201. [DOD] [CrossRef]
- Reichen J. The role of the sinusoidal endothelium in liver function. Physiology 1999. 14(3):117-121. [DOD]
- Polonsky KS, Given BD, Hirsch L, Shapiro ET, Tillil H, Beebe C, Galloway JA, Frank BH, Karrison T, Van Cauter E. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest 1988. 81:435-441. [DOD] [CrossRef]
- Waldhausl W, Bratusch-Marrain P, Gasic S, Korn A, Nowotny P. Insulin production rate following glucose ingestion estimated by splanchnic C-peptide output in normal man. Diabetologia 1979. 17(4):221-227. [DOD] [CrossRef]
- Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev 1998. 19(5):608-624. [DOD] [CrossRef]
- White MF, Kahn CR. The insulin signaling system. J Biol Chem 1994. 269(1):1-4. [DOD]
- Authier F, Posner BI, Bergeron JJ. Insulin-degrading enzyme. Clin Invest Med 1996. 19(3):149-160. [DOD]
- Akiyama H, Yokono K, Shii K, Ogawa W, Taniguchi H, Baba S, Kasuga M. Natural regulatory mechanisms of insulin degradation by insulin degrading enzyme. Biochem Biophys Res Commun 1990. 170(3):1325-1330. [DOD] [CrossRef]
- Pivovarova O, Gögebakan Ö, Pfeiffer AF, Rudovich N. Glucose inhibits the insulin-induced activation of the insulin-degrading enzyme in HepG2 cell. Diabetologia 2009. 52:1656-1664. [DOD] [CrossRef]
- Rorsman P, Eliasson L, Renström E, Gromada J, Barg S, Göpel S. The cell physiology of biphasic insulin secretion. News Physiol Sci 2000. 15:72-77. [DOD]
- Schmitz O, Rungby J, Edge L, Juhl CB. On high-frequency insulin oscillations. Ageing Res Rev 2008. 7(4):301-305. [DOD] [CrossRef]
- Hellman B, Gylfe E, Bergsten P, Grapengiesser E, Lund PE, Berts A, Tengholm A, Pipeleers DG, Ling Z. Glucose induces oscillatory Ca2+ signalling and insulin release in human pancreatic beta cells. Diabetologia 1994. 37(Suppl 2):11-20. [DOD] [CrossRef]
- Porksen N. The in vivo regulation of pulsatile insulin secretion. Diabetologia 2002. 45(1):3-20. [DOD] [CrossRef]
- Gylfe E, Grapengiesser E, Hellman B. Propagation of cytoplasmic Ca2+ oscillations in clusters of pancreatic (beta)-cells exposed to glucose. Cell Calcium 1991. 12(2-3):229-240. [DOD] [CrossRef]
- Ravier MA, Güldenagel M, Charollais A, Gjinovci A, Caille D, Söhl G, Wollheim CB, Willecke K, Henquin JC, Meda P. Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 2005. 54(6):1798-1807. [DOD] [CrossRef]
- Gilon P, Ravier MA, Jonas JC, Henique JC. Control mechanisms of the oscillations of insulin secretion in vitro and in vivo. Diabetes 2002. 51(Suppl 1):S144-S151. [DOD] [CrossRef]
- Porksen N, Munn S, Ferguson D, O'Brien T, Veldhuis JD, Butler P. Coordinate pulsatile insulin secretion by chronic intraportally transplanted islets in the isolated perfused rat liver. J Clin Invest 1994. 94(1):219-227. [DOD] [CrossRef]
- Simon C, Brandenberger G. Ultradian oscillations of insulin secretion in humans. Diabetes 2002. 51(Suppl 1):S258-S261. [DOD] [CrossRef]
- Bergsten P. Pathophysiology of impaired pulsatile insulin release. Diabetes Metab Res Rev 2000. 16(3):179-191. [DOD] [CrossRef]
- Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 2001. 358(9277):221-229. [DOD] [CrossRef]
- Czech MP. The nature and regulation of the insulin receptor: Structure and function. Ann Rev Physiol 1985. 47:357-381. [DOD] [CrossRef]
- Kahn CR, White MF. The insulin receptor and the molecular mechanism of insulin action. J Clin Invest 1988. 82(4):1151-1156. [DOD] [CrossRef]
- Kasuga M, Karlsson FA, Kahn CR. Insulin stimulates the phosphorylation of the 95,000 dalton subunit of its own receptor. Science 1982. 215:185-187. [DOD] [CrossRef]
- White MF. The insulin signalling system and the IRS proteins. Diabetologia 1997. 40(Suppl 2):S2-S17. [DOD] [CrossRef]
- Gonzalez FA, Seth A, Raden DL, Bowman DS, Fay FS, Davis RJ. Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J Cell Biol 1993. 122(5):1089-1101. [DOD] [CrossRef]
- Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J 1998. 333(Pt 3):471-490. [DOD]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004. 166(2):213-223. [DOD] [CrossRef]
- Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 2002. 3(4):267-277. [DOD] [CrossRef]
- Proud CG. Regulation of protein synthesis by insulin. Biochem Soc Trans 2006. 34(Pt 2):213-216. [DOD]
- Thorens B, Sarkar HK, Kaback HR, Lodish HF. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988. 55:281-290. [DOD]
- Rencurel F, Waeber G, Antoine B, Rocchiccioli F, Maulard P, Girard J, Leturque A. Requirement of glucose metabolism for regulation of glucose transporter type 2 (GLUT2) gene expression in liver. Biochem J 1996. 314(3):903-909. [DOD]
- Asano T, Katagiri H, Tsukuda K, Lin LJ, Ishihara H, Yazaki Y, Oka Y. Upregulation of GLUT2 mRNA by glucose, mannose, and fructose in isolated rat hepatocytes. Diabetes 1992. 41:22-25. [DOD] [CrossRef]
- Postic C, Burcelin R, Rencurel F, Pegorier JP, Loizeau M, Girard J, Leturque A. Evidence for a transient inhibitory effect of insulin on GLUT2 expression in the liver: studies in vivo and in vitro. Biochem J 1993. 293(1):119-124. [DOD]
- Thorens B, Flier JS, Lodish HF, Kahn BB. Differential regulation of two glucose transporters in rat liver by fasting and refeeding and by diabetes and insulin treatment. Diabetes 1990. 39:712-719. [DOD] [CrossRef]
- Burcelin R, Eddouks M, Kande J, Assan R, Girard J. Evidence that GLUT-2 mRNA and protein concentrations are decreased by hyperinsulinaemia and increased by hyperglycaemia in liver of diabetic rats. Biochem J 1992. 288(2):675-679. [DOD]
- Barthel A, Schmoll D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am J Physiol Endo 2003. 285(4):E685-E692. [DOD]
- Gold AH. The effect of diabetes and insulin on liver glycogen synthetase activation. J Biol Chem 1970. 245(4):903-905. [DOD]
- Flatt JP. Use and storage of carbohydrate and fat. Am J Clin Nutr 1995. 61(4):952S-959S. [DOD]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002. 109(9):1125-1131. [DOD]
- Azzout-Marniche D, Becard D, Guichard C, Foretz M, Ferre P, Foufelle F. Insulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes. Biochem J 2000. 350(2):389-393. [DOD] [CrossRef]
- Im SS, Kang SY, Kim SY, Kim HI, Kim JW, Kim KS, Ahn YH. Glucose-stimulated upregulation of GLUT2 gene is mediated by sterol response element-binding protein-1c in the hepatocytes. Diabetes 2005. 54(6):1684-1691. [DOD] [CrossRef]
- Ferre P, Foufelle F. SREBP-1c Transcription Factor and Lipid Homeostasis: Clinical Perspective. Horm Res 2007. 68:72-82. [DOD] [CrossRef]
- Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004. 114(2):147-152. [DOD]
- Weickert MO, Pfeiffer AF. Signalling mechanisms linking hepatic glucose and lipid metabolism. Diabetologia 2006. 49(8):1732-1741. [DOD] [CrossRef]
- Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol 1999. 276(1 Pt 1):E1-E18. [DOD]
- Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, Arnot D, Uyeda K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci USA 2001. 98(16):9116-9121. [DOD] [CrossRef]
- Elasha H, Alzahrani S, Almalki M, Alomair A, Alshakweer W. Acute hepatic steatosis: an unusual manifestation of poorly controlled type 1 diabetes. Pract Diab Int 2010. 27(7):282-283. [DOD] [CrossRef]
- Tobin KA, Ulven SM, Schuster GU, Steineger HH, Andresen SM, Gustafsson JA, Nebb HI. Liver X receptors as insulin-mediating factors in fatty acid and cholesterol biosynthesis. J Biol Chem 2002. 277:10691-10697. [DOD] [CrossRef]
- Wong RH, Sul HS. Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr Opin Pharmacol 2010. 10(6):684-691. [DOD] [CrossRef]
- Casado M, Vallet VS, Kahn A, Vaulont S. Essential role in vivo of upstream stimulatory factors for a normal dietary response of the fatty acid synthase gene in the liver. J Biol Chem 1999. 274(4):2009-2013. [DOD] [CrossRef]
- Griffin MJ, Wong RH, Pandya N, Sul HS. Direct interaction between USF and SREBP-1c mediates synergistic activation of the fatty-acid synthase promoter. J Biol Chem 2007. 282(8):5453-5467. [DOD] [CrossRef]
- Softic S, Kirby M, Shroyer N, Kohli R. Hepatic steatosis in type 2 and type 1 diabetes mellitus is mediated by insulin signaling via fatty acid transport proteins. J Pediatr Gastroenterol Nutr 2010. 51(Suppl 2):E33-E34. [DOD]
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002. 346(16):1221-1231. [DOD] [CrossRef]
- Tominaga K, Kurata JH, Chen YK, Fujimoto E, Miyagawa S, Abe I, Kusano Y. Prevalence of fatty liver in Japanese children and relationship to obesity. An epidemiological ultrasonographic survey. Dig Dis Sci 1995. 40(9):2002-2009. [DOD] [CrossRef]
- Franzese A, Vajro P, Argenziano A, Puzziello A, Iannucci MP, Saviano MC. Liver involvement in obese children. Ultrasonography and liver enzyme levels at diagnosis and during follow-up in an Italian population. Dig Dis Sci 1997. 42(7):1428-1432. [DOD] [CrossRef]
- Kotronen A, Vehkavaara S, Seppälä-Lindroos A, Bergholm R, Yki-Järvinen H. Effect of liver fat on insulin clearance. Am J Physiol Endo 2007. 293(6):E1709-E1715. [DOD]
- Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, Goto T, Westerbacka J, Sovijarvi A, Halavaara J, Yki-Jarvinen H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 2002. 87:3023-3028. [DOD] [CrossRef]
- Ryysy L, Häkkinen AM, Goto T, Vehkavaara S, Westerbacka J, Halavaara J, Yki-Järvinen H. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 2000. 49:749-758. [DOD] [CrossRef]
- Juurinen L, Tiikainen M, Häkkinen AM, Hakkarainen A, Yki-Järvinen H. Effects of insulin therapy on liver fat content and hepatic insulin sensitivity in patients with type 2 diabetes. Am J Physiol Endo 2007. 292(3):E829-E835. [DOD]
- Neuschwander-Tetri BA. Fatty liver and nonalcoholic steatohepatitis. Clincal cornerstone 2001. 3(6):47-57. [DOD] [CrossRef]
- Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med 2001. 344:495-500. [DOD] [CrossRef]
- Rashid M, Roberts EA. Nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2000. 30(1):48-53. [DOD] [CrossRef]
- Manton ND, Lipsett J, Moore DJ, Davidson GP, Bourne AJ, Couper RT. Non-alcoholic steatohepatitis in children and adolescents. Med J Aust 2000. 173(9):476-479. [DOD]
- Al-Hussaini AA, Sulaiman N, Al-Zahrani M, Alenazi A, Khan M. Prevalence of liver disease among type 1 diabetic children. J Pediatr Gastroenterol Nutr 2010. 51(Suppl 2):E57. [DOD]
- El-Karaksy HM, Anwar G, Esmat G, Mansour S, Sabry M, Helmy H, El-Hennawy A, Fouad H. Prevalence of hepatic abnormalities in a cohort of Egyptian children with type 1 diabetes mellitus. Pediatr Diabetes 2010. 11(7):462-470. [DOD] [CrossRef]
- Gaiani S, Avogaro A, Bombonato GC, Bolognesi M, Amor F, Vigili de Kreutzenberg S, Guarneri G, Sacerdoti D. Nonalcoholic fatty liver disease (NAFLD) in nonobese patients with diabetes: Prevalence and relationships with hemodynamic alterations detected with Doppler sonography. J Ultrasound 2009. 12(1):1-5. [DOD] [CrossRef]
- Targher G, Bertolini L, Padovani R, Rodella S, Zoppini G, Pichiri I, Sorgato C, Zenari L, Bonora E. Prevalence of non-alcoholic fatty liver disease and its association with cardiovascular disease in patients with type 1 diabetes. J Hepatol 2010. 53(4):713-718. [DOD] [CrossRef]
- Targher G, Pichiri I, Zoppini G, Trombetta M, Bonora E. Increased prevalence of chronic kidney disease in patients with type 1 diabetes and non-alcoholic fatty liver. Diabet Med 2012. 29(2):220-226. [DOD] [CrossRef]
- Saadeh S, Younossi ZM, Remer EM, Gramlich T, Ong JP, Hurley M, Mullen KD, Cooper JN, Sheridan MJ. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology 2002. 123(3):745-750. [DOD] [CrossRef]
- Pozzato C, Radaelli G, Dall'Asta C, Verduci E, Villa A, Villa C, Scaglioni S, Riva E, Pontiroli AE, Cornalba G, et al. MRI in identifying hepatic steatosis in obese children and relation to ultrasonography and metabolic findings. J Pediatr Gastroenterol Nutr 2008. 47(4):493-499. [DOD]
- Kilpatrick ES, Rigby AS, Atkin SL. Insulin resistance, the metabolic syndrome, and complication risk in type 1 diabetes: "double diabetes" in the Diabetes Control and Complications Trail. Diabetes Care 2007. 30:707-712. [DOD] [CrossRef]
- Thorn LM, Forsblom C, Fagerudd J, Thomas MC, Pettersson-Fernholm K, Saraheimo M. Metabolic syndrome in type 1 diabetes: association with diabetic nephropathy and glycemic control (the FinnDiane study). Diabetes Care 2005. 28(8):2019-2024. [DOD] [CrossRef]
- Pambianco G, Costacou T, Orchyard TJ. The prediction of major outcomes of type 1 diabetes: a 12-year prospective study of three separate definitions of the metabolic syndrome and their components and estimated glucose disposal rate. The Pittsburgh Epidemiology of Diabetes Complications Study Experience. Diabetes Care 2007. 30:1248-1254. [DOD] [CrossRef]
- Alberti KG, Zimmet P, Shaw J. Metabolic syndrome: a new world-wide definition: a consensus statement from the International Diabetes Federation. Diabet Med 2006. 23:469-480. [DOD] [CrossRef]
- DeFronzo RA, Simonson D, Ferrannini E. Hepatic and peripheral insulin resistance: a common feature of type 2 (non-insulin-dependent) and type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1982. 23:313-319. [DOD]
- Yki-Jarvinen H. Fat in the liver and insulin resistance. Ann Med 2005. 37(5):347-356. [DOD] [CrossRef]
- Schindhelm RK, Diamant M, Dekker JM, Tushuizen ME, Teerlink T, Heine RJ. Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes Metab Res Rev 2006. 22(6):437-443. [DOD] [CrossRef]
- Adams LA, Angulo P, Lindor KD. Nonalcoholic fatty liver disease. CMAJ 2005. 172(7):899-905. [DOD] [CrossRef]
- Mofrad P, Contos MJ, Haquw M, Sargeant C, Fisher RA, Luketic VA. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003. 37(6):1286-1292. [DOD] [CrossRef]
- West J, Brousil J, Gazis A, Jackson L, Mansell P, Bennett A. Elevated serum alanine transaminase in patients with type 1 or type 2 diabetes mellitus. QJM 2006. 99(12):871-876. [DOD] [CrossRef]
- Leeds JS, Forman EM, Morely S, Scott AR, Tesfaye S, Sanders DS. Abnormal liver function tests in patients with type 1 diabetes mellitus: prevalence, clinical correlations and underlying pathologies. Diabet Med 2009. 26:1235-1241. [DOD] [CrossRef]
- Bulum T, Kolaric B, Duvnjak L, Duvnjak M. Nonalcoholic fatty liver disease markers are associated with insulin resistance in type 1 diabetes. Dig Dis Sci 2011. 56(12):3655-3663. [DOD] [CrossRef]
- Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, Nierras CR, Todd JA, Rich SS, Nerup J. Genetics of type 1 diabetes: what's next? Diabetes 2010. 59(7):1561-1571. [DOD]
- Melendez-Ramirez LY, Richards RJ, Cefalu WT. Complications of type 1 diabetes. Endocrinol Metab Clin North Am 2010. 39(3):625-640. [DOD] [CrossRef]
- Targher G, Pichiri I, Zoppini G, Trombetta M, Bonora E. Increased prevalence of cardiovascular disease in type 1 diabetic patients with non-alcoholic fatty liver disease. J Endocrinol Invest 2011. In press. [DOD]
- Targher G, Bertolini L, Chonchol M, Rodella S, Zoppini G, Lippi G, Zenari L, Bonora E. Non-alcoholic fatty liver disease is independently associated with an increased prevalence of chronic kidney disease and retinopathy in type 1 diabetic patients. Diabetologia 2010. 53(7):1341-1348. [DOD] [CrossRef]
- Verges B. Lipid disorders in type 1 diabetes. Diabetes Metab 2009. 35(5):353-360. [DOD] [CrossRef]
- Lenaerts J, Verresen L, Van Steenbergen W, Fevery J. Fatty liver hepatitis and type 5 hyperlipoproteinemia in juvenile diabetes mellitus. Case report and review of the literature. J Clin Gastroenterol 1990. 12(1):93-97. [DOD] [CrossRef]
- Roden M, Bernroider E. Hepatic glucose metabolism in humans - its role in health and disease. Best Pract Res Clin Endocrinol Metab 2003. 17(3):365-383. [DOD] [CrossRef]
- Cline GW, Rothman DL, Magnusson I, Katz LD, Shulman GI. 13C-nuclear magnetic resonance spectroscopy studies of hepatic glucose metabolism in normal subjects and subjects with insulin-dependent diabetes mellitus. J Clin Invest 1994. 94(6):2369-2376. [DOD] [CrossRef]
- Bischof MG, Krssak M, Krebs M, Bernroider E, Stingl H, Waldhäusl W, Roden M. Effects of short-term improvement of insulin treatment and glycemia on hepatic glycogen metabolism in type 1 diabetes. Diabetes 2001. 50(2):392-398. [DOD] [CrossRef]
- Hwang JH, Perseghin G, Rothman DL, Cline GW, Magnusson I, Petersen KF. Impaired net hepatic glycogen synthesis in insulin-dependent diabetic subjects during mixed meal ingestion. A 13C nuclear magnetic resonance spectroscopy study. J Clin Invest 1995. 95(2):783-787. [DOD] [CrossRef]
- Matyka K, Dixon RM, Mohn A, Rajagopalan B, Shmueli E, Styles P, Dunger DB. Daytime liver glycogen accumulation, measured by 13C magnetic resonance spectroscopy, in young children with Type 1 diabetes mellitus. Diabet Med 2001. 18(8):659-662. [DOD] [CrossRef]
- Petersen KF, Laurent D, Rothman DL, Cline GW, Shulman GI. Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J Clin Invest 1998. 101(6):1203-1209. [DOD] [CrossRef]
- Kishore P, Gabriely I, Cui MH, Di Vito J, Gajavelli S, Hwang JH, Shamoon H. Role of hepatic glycogen breakdown in defective counterregulation of hypoglycemia in intensively treated type 1 diabetes. Diabetes 2006. 55(3):659-666. [DOD] [CrossRef]
- Bronstein HD, Kantrowitz PA, Schaffner F. Marked enlargement of the liver and transient ascites associated with the treatment of diabetic acidosis. N Engl J Med 1959. 261:1314-1318. [DOD] [CrossRef]
- Munns CF, McCrossin RB, Thomsett MJ, Batch J. Hepatic glycogenosis: reversible hepatomegaly in type 1 diabetes. J Paediatr Child Health 2000. 36(5):449-452. [DOD] [CrossRef]
- Torbenson M, Chen YY, Brunt E, Cummings OW, Gottfried M, Jakate S. Glycogenic hepatopathy: an underrecognized hepatic complication of diabetes mellitus. Am J Surg Pathol 2006. 30(4):508-513. [DOD] [CrossRef]
- Aljabri KS, Bokhari SA, Fageeh SM, Alharbi AM, Abaza MA. Glycogen hepatopathy in a 13-year-old male with type 1 diabetes. Ann Saudi Med 2011. 31(4):424-427. [DOD] [CrossRef]
- Nakamuta M, Ohashi M, Goto K, Tanabe Y, Hiroshige K, Nawata H. Diabetes mellitus-associated glycogen storage hepatomegaly: report of a case and review of the Japanese literature. Fukuoka Igaku Zasshi 1993. 84(7):354-358. [DOD]
- Olsson R, Wesslau C, William-Olsson T, Zettergren L. Elevated aminotransferases and alkaline phosphatases in unstable diabetes mellitus without ketoacidosis or hypoglycemia. J Clin Gastroenterol 1989. 11(5):541-545. [DOD] [CrossRef]
- Carcione L, Lombardo F, Messina MF, Rosano M, De Luca F. Liver glycogenosis as early manifestation in type 1 diabetes mellitus. Diabetes Nutr Metab 2003. 16(3):182-184. [DOD]
- Vallance-Owen J. Liver glycogen in diabetes mellitus. J Clin Pathol 1952. 5(1):42-53. [DOD] [CrossRef]
- Evans RW, Littler TR, Pemberton HS. Glycogen storage in the liver in diabetes mellitus. J Clin Pathol 1955. 8(2):110-113. [DOD] [CrossRef]
- Abaci A, Bekem O, Unuvar T, Ozer E, Bober E, Arslan N. Hepatic glycogenosis: a rare cause of hepatomegaly in type 1 diabetes mellitus. J Diabetes Complications 2008. 22(5):325-328. [DOD] [CrossRef]
- Chatila R, West AB. Hepatomegaly and abnormal liver tests due to glycogenosis in adults with diabetes. Medicine (Baltimore) 1996. 75(6):327-333. [DOD] [CrossRef]
- Unger RH. Role of glucagon in the pathogenesis of diabetes: the status of the controversy. Metabolism 1978. 27(11):1691-1709. [DOD] [CrossRef]
- Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 2005. 54(6):1789-1797. [DOD] [CrossRef]
- Aguilar-Parada E, Eisentraut AM, Unger RH. Pancreatic glucagon secretion in normal and diabetic subjects. Am J Med Sci 1969. 257(6):415-419. [DOD] [CrossRef]
- Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA 2010. 107(37):16009-16012. [DOD] [CrossRef]
- Ramnanan CJ, Edgerton DS, Kraft G, Cherrington AD. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab 2011. 13(Suppl 1):118-125. [DOD] [CrossRef]
- Bewsher PD, Ashmore J. Ketogenic and lipolytic effects of glucagon on liver. Biochem Biophys Res Commun 1966. 24(3):431-436. [DOD] [CrossRef]
- Liljenquist JE, Bomboy JD, Lewis SB, Sinclair-Smith BC, Felts PW, Lacy WW. Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men. J Clin Invest 1974. 53(1):190-197. [DOD] [CrossRef]
- Berglund ED, Lustig DG, Baheza RA, Hasenour CM, Lee-Young RS, Donahue EP, Lynes SE, Swift LL, Charron MJ, Damon BM, et al. Hepatic glucagon action is essential for exercise-induced reversal of mouse fatty liver. Diabetes 2011. 60(11):2720-2729. [DOD] [CrossRef]
- Kolterman OG, Schwartz S, Corder C, Levy B, Klaff L, Peterson J. Effect of 14 days' subcutaneous administration of the human amylin analogue, pramlintide (AC137), on an intravenous insulin challenge and response to a standard liquid meal in patients with IDDM. Diabetologia 1996. 39(4):492-499. [DOD] [CrossRef]
- Fineman M, Weyer C, Maggs DG, Strobel S, Kolterman OG. The human amylin analog, pramlintide, reduces postprandial hyperglucagonemia in patients with type 2 diabetes mellitus. Horm Metab Res 2002. 34:504-508. [DOD] [CrossRef]
- Polonsky KS, Rubenstein AH. C-peptide as a measure of the secretion and hepatic extraction of insulin. Pitfalls and limitations. Diabetes 1984. 33:486-494. [DOD] [CrossRef]
- Wahren J. C-peptide: new findings and therapeutic implications in diabetes. Clin Physiol Funct Imaging 2004. 24(4):180-189. [DOD] [CrossRef]
- Hills CE, Brunskill NJ. C-Peptide and its intracellular signaling. Rev Diabet Stud 2009. 6(3):138-147. [DOD] [CrossRef]
- Lonovics J, Devitt P, Watson LC, Rayford PL, Thompson JC. Pancreatic polypeptide. A review. Arch Surg 1981. 116(10):1256-1264. [DOD] [CrossRef]
- Schwartz TW. Pancreatic polypeptide: a unique model for vagal control of endocrine systems. J Auton Nerv Syst 1983. 9(1):99-111. [DOD] [CrossRef]
- Loba JM, Saryusz-Wolska M, Czupryniak L, Kukulski K. Pancreatic polypeptide secretion in diabetic patients with delayed gastric emptying and autonomic neuropathy. J Diabetes Complications 1997. 11(6):328-333. [DOD] [CrossRef]
- Skare S, Hanssen KF, Lundqvist G. Increased plasma pancreatic polypeptide (PP) in diabetic ketoacidosis; normalization following treatment. Acta Endocrinol (Copenh) 1980. 93(4):466-469. [DOD]
- Nguyen TD, MS Wolfe, Heintz GG, Whitcomb DC, Taylor IL. High affinity binding proteins for pancreatic polypeptide on rat liver membranes. J Biol Chem 1992. 267(13): 9416-9421. [DOD]
- Seymour NE, Volpert AR, Andersen DK. Regulation of hepatic insulin receptors by pancreatic polypeptide in fasting and feeding. J Surg Res 1996. 65(1):1-4. [DOD] [CrossRef]
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000. 45(10):1929-1934. [DOD] [CrossRef]
- Targher F, Bertolini L, Padovani R, Rodella S, Tessari R, Zenari L, Day C, Arcaro G. Prevalence of nonalcoholic fatty liver disease and Its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007. 30(5):1212-1218. [DOD] [CrossRef]
- Williamson RM, Price JF, Glancy S, Perry E, Nee LD, Hayes PC, Frier BM, Van Look LA, Johnston GI, Reynolds RM, et al. Prevalence of and risk factors for hepatic steatosis and nonalcoholic fatty liver disease in people with type 2 diabetes: the Edinburgh Type 2 Diabetes Study. Diabetes Care 2011. 34(5):1139-1144. [DOD] [CrossRef]
- Mennesson N, Dumortier J, Hervieu V, Milot L, Guillaud O, Scoazec JY, Pilleul F. Liver steatosis quantification using magnetic resonance imaging: a prospective comparative study with liver biopsy. J Comput Assist Tomogr 2009. 33(5):672-677. [DOD] [CrossRef]
- Springer F, Machann J, Claussen CD, Schick F, Schwenzer NF. Liver fat content determines by magnetic resonance imaging and spectroscopy. World J Gastroenterol 2010. 16(13):1560-1566. [DOD] [CrossRef]
- Lecomte M, Gottrand F, Stuckens C, Lecomte-Houcke M. Acute steatosis in an 8-year-old boy with insulin-dependent diabetes mellitus. J Pediatr Gastroenterol Nutr 1997. 25(1):98-100. [DOD] [CrossRef]
- Mehta SR, Hameed A, Theofanoyiannis P, Robinson S, Valabhji J, Elkeles RS. Non-alcoholic steatohepatitis (NASH) in a patient with type 1 diabetes. Endocrine Abstracts 2005. 10:21. [DOD]
This article has been cited by other articles:
|
Hepatic expression of the Sptlc3 subunit of serine palmitoyltransferase is associated with the development of hepatocellular carcinoma in a mouse model of nonalcoholic steatohepatitis
Yoshimine Y, Uto H, Kumagai K, Mawatari S, Arima S, Ibusuki R, Mera K, Nosaki T, Kanmura S, Numata M, Tamai T, Moriuchi A, Tsubouchi H, Ido A
Oncol Rep 2015. 33(4):1657-1666
|
|

|
Effect of exercise training on liver antioxidant enzymes in STZ-diabetic rats
Lima TI, Monteiro IC, Valença S, Leal-Cardoso JH, Fortunato RS, Carvalho DP, Teodoro BG, Ceccatto VM
Life Sci 2015. pii:S0024-3205(15)00100-9
|
|

|
Alloxan-Induced Diabetes Causes Morphological and Ultrastructural Changes in Rat Liver that Resemble the Natural History of Chronic Fatty Liver Disease in Humans
Lucchesi AN, Cassettari LL, Spadella CT
J Diabetes Res 2015. 2015:494578
|
|

|
Clinical profile of nonalcoholic fatty liver disease among young patients with type 1 diabetes mellitus seen at a diabetes speciality center in India
Vendhan R, Amutha A, Anjana RM, Unnikrishnan R, Mohan V
Endocr Pract 2014. 20(12):1249-1257
|
|

|
Effects of sodium-glucose cotransporter 2 selective inhibitor ipragliflozin on hyperglycaemia, oxidative stress, inflammation and liver injury in streptozotocin-induced type 1 diabetic rats
Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, Takasu T, Imamura M, Li Q, Tomiyama H, Kobayashi Y, Noda A, Sasamata M, Shibasaki M
J Pharm Pharmacol 2014. 66(7):975-987
|
|

|
Prevalence and natural history of histologically proven chronic liver disease in a longitudinal cohort of patients with type 1 diabetes
Harman DJ, Kaye PV, Harris R, Suzuki A, Gazis A, Aithal GP
Hepatology 2014. 60(1):158-168
|
|

|
Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes
Sahini N, Borlak J
Prog Lipid Res 2014. 54:86-112
|
|

|
Passive fructose transporters in disease: a molecular overview of their structural specificity
McQuade DT, Plutschack MB, Seeberger PH
Org Biomol Chem 2013. 11(30):4909-4920
|
|

|
Glycogen storage disease type 1 and diabetes: learning by comparing and contrasting the two disorders
Rajas F, Labrune P, Mithieux G
Diabetes Metab 2013. 39(5):377-387
|
|

|
Cannabinoid 1 receptor in fatty liver
Regnell SE
Hepatol Res 2013. 43(2):131-138
|
|

|
Non-alcoholic fatty liver disease in common endocrine disorders
Hazlehurst JM, Tomlinson JW
Eur J Endocrinol 2013. 169(2):R27-R37
|
|

|
The role of fructose transporters in diseases linked to excessive fructose intake
Douard V, Ferraris RP
J Physiol 2013. 591(Pt 2):401-414
|
|

|
Nonalcoholic fatty liver disease: an emerging threat to obese and diabetic individuals
Masuoka HC, Chalasani N
Ann N Y Acad Sci 2013. 1281:106-122
|
|
|
Flow synthesis of a versatile fructosamine mimic and quenching studies of a fructose transport probe
Plutschack MB, McQuade DT, Valenti G, Seeberger PH
Beilstein J Org Chem 2013. 9:2022-2027
|
|
|


)
)
)
)

