Review
| Rev Diabet Stud,
2007,
4(3):134-146 |
DOI 10.1900/RDS.2007.4.134 |
Adipose Tissue: A Metabolic Regulator. Potential Implications for the Metabolic Outcome of Subjects Born Small for Gestational Age (SGA)
Arianna Maiorana, Chiara Del Bianco, Stefano Cianfarani
Rina Balducci Center of Pediatric Endocrinology, Department of Public Health and Cell Biology, Tor Vergata University, 00133-Rome, Italy
Address correspondence to: Stefano Cianfarani, e-mail: stefano.cianfarani@uniroma2.it
Keywords: diabetes, SGA, adipose tissue, glucose metabolism, lipids, insulin resistance, low birth weight
Abstract
Adipose tissue is involved in the regulation of glucose and lipid metabolism, energy balance, inflammation and immune response. Abdominal obesity plays a key role in the development of insulin resistance because of the high lipolytic rate of visceral adipose tissue and its secretion of adipocytokines. Low birth weight subjects are prone to central redistribution of adipose tissue and are at high risk of developing metabolic syndrome, type 2 diabetes and cardiovascular disease. Intrauterine adipogenesis may play a key role in the fetal origin of the pathogenesis of metabolic syndrome, type 2 diabetes and cardiovascular disease. Therefore, knowledge of the behavior of visceral adipose tissue-derived stem cells could provide a greater understanding of the metabolic risk related to intrauterine growth retardation, with potential clinical implications for the prevention of long-term metabolic alterations.
Adipose tissue and insulin resistance
Pathophysiology of visceral adipose tissue: the role of free fatty acids
Adipocytes are highly specialized cells that maintain whole body energy homeostasis by regulating glucose and lipid metabolism. For a long time, adipocytes have been considered to be an energy depot that stores and mobilizes triglycerides [1]. Recently, adipocytes have also been recognized as an endocrine tissue because of the discovery of several adipocyte-derived molecules, including lipid metabolites and adipocytokines [2].
Adipose tissue contains functionally distinct cellular subtypes, the white adipocyte, devoted to energy storage, and the brown adipocyte, which dissipates energy through thermogenesis. In the rat, brown adipose tissue is the major site of heat production. Brown fat also plays a major role in heat production in the neonates of many mammalian species. Its quantitative contribution to energy metabolism at maturity in large mammals, including humans, is uncertain. The storage of triglycerides and fatty acids in white adipose tissue occurs through the ability of insulin to stimulate significantly both glucose uptake and lipogenesis. Defects in fuel partitioning into adipocytes either because of increased adipose mass or increased circulating free fatty acids, results in dyslipidemia, obesity, insulin resistance and type 2 diabetes [3].
The association between type 2 diabetes and obesity is well established. Several studies have documented that the incidence of diabetes rises steeply with increasing body weight [4-6]. The parallel rapid growth of overweight in obese individuals and type 2 diabetes is striking, and has led to the coining of the term ‘diabesity’ to emphasize the link between these two conditions [7]. Obesity is an insulin resistance state, and both obesity and insulin resistance are risk factors for the development of type 2 diabetes [8, 9]. In addition, the pattern of fat distribution is also an important predictor of insulin sensitivity. Individuals with preferential upper body fat accumulation (android) are more insulin resistant, hyperinsulinemic and dyslipidemic than people with a preponderance of lower body fat (gynecoid) [10, 11].
Increased visceral fat has been shown to be specifically related to insulin resistance. This association has been attributed to the enhanced lipolytic activity of visceral fat cells, with increased delivery of free fatty acids (FFA) into the portal (causing hepatic insulin resistance) and systemic (causing muscle insulin resistance) circulation [12, 13]. Patients with type 2 diabetes are characterized by day-long elevation in plasma FFA concentration, which is not suppressed after meals [14] or in response to insulin [15]. Chronically elevated plasma FFA concentrations cause insulin resistance in muscle and liver [15-17] and impair insulin secretion [18, 19] (Figure 1). Furthermore, depots of fatty acids stored in the form of triglycerides have been found in the muscle [20, 21] and liver [22, 23] of type 2 diabetic patients. The adipocytes of subjects with type 2 diabetes are enlarged cells with diminished capacity to store fat. When adipocyte storage capacity is exceeded, lipids 'overflow' into muscle and liver and possibly into the β-cells of the pancreas [8]. This ectopic fat deposition in muscle and liver has been referred to as one element of lipotoxicity [18, 24-26].
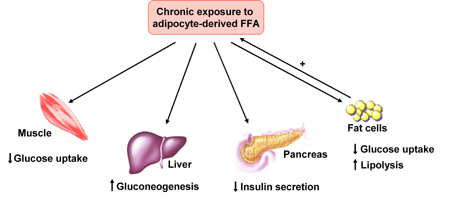 |
 |
Figure 1. Action of adipocyte-derived free fatty acids (FFA) on glucose homeostasis. FFAs induce muscle insulin resistance by inhibiting insulin-mediated glucose uptake, and hepatic insulin resistance by affecting insulin-mediated suppression of hepatic glucose output. Chronic exposure to FFAs impairs insulin secretion from β-cells and, in the adipose tissue, reduces insulin-mediated glucose uptake and increases lipolysis. Modified from [44]. |
|
Muscle insulin resistance results in decreased glucose uptake [27] and consequently in impaired glucose oxidation and glycogen synthesis [8, 28]. In the liver, insulin resistance results in the impaired ability of insulin to suppress hepatic glucose production (HGP) [27-29] (Figure 2).
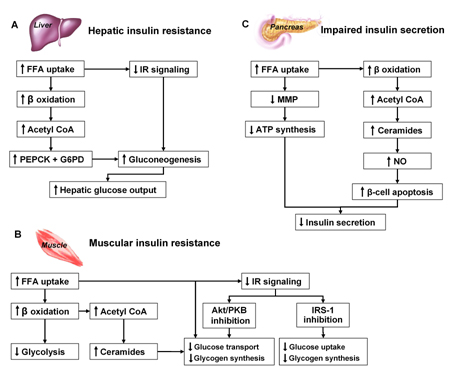 |
|
Figure 2. FFA action on liver (A), muscle (B) and β-cells (C). A: In the liver, FFAs increase lipid oxidation and accumulation of acetyl CoA. Acetyl CoA stimulates the rate-limiting enzymes for gluconeogenesis eventually leading to increased glucose output. Finally, elevated plasma FFAs inhibit the insulin signal transduction system [24, 113, 114]. B: In muscle, FFAs reduce glucose oxidation by affecting the redox potential of myocytes and inhibiting key glycolytic enzymes. Furthermore, FFAs increase IRS-1 serine phosphorylation, through activation of protein kinase C, thus inhibiting insulin signaling. Finally, ceramide accumulation interferes with glucose transport and inhibits glycogen synthesis via inhibition of protein kinase B [24, 115-117]. C: In the pancreas, lipotoxicity is an important cause of β-cell dysfunction [18, 24, 118]. If short-term elevation (e.g. after a meal) of plasma FFAs enhances insulin secretion [119], long term exposure to FFAs (>48 hours) impairs insulin secretion and eventually leads to β-cells apoptosis [120, 121]. Increased formation of ceramides from accumulated acetyl CoA augments the nitric oxide formation that causes apoptosis of β-cells [118]. This has been demonstrated in vitro in human β-cells incubated with FFAs [122], and in vivo in rodents [123]. FFA: free fatty acids. IR: insulin receptor. PEPCK: phosphoenolpyruvate carboxyl kinase. G6PD: glucose-6-phosphatase. NO: nitric oxide. Acetyl CoA: acetyl coenzyme A. PKB/Akt: protein kinase B. IRS-1: insulin receptor substrate-1. MMP: mitochondrial membrane potential. |
|
Increased intramyocellular triglyceride content is associated with impaired glucose tolerance and type 2 diabetes and is a better predictor of impaired insulin action than visceral adiposity [30]. Ectopic fat deposition leads to insulin resistance, as the intracellular metabolites of triglyceride metabolism interfere with insulin signaling, glucose transport and glycogen synthesis in muscle, and augment hepatic gluconeogenesis. In cases of dysfunctional adipose tissue, which is insulin resistant with a limited ability to store the energy excess, the triglyceride surplus is deposited in liver, skeletal muscle, heart and visceral adipose tissue [7].
Subjects born small for gestational age (SGA) are prone to central redistribution of adipose tissue and are at high risk of developing insulin resistance, type 2 diabetes, metabolic syndrome and cardiovascular disease [31].
The role of adipocytokines
Adipose tissue produces a wide variety of signaling molecules involved in inflammation, food intake and insulin sensitivity, which are known as adipocytokines. In type 2 diabetes, this adipocyte function is defective: fat cells, particularly from visceral adipose tissue, produce excessive amounts of proinflammatory, insulin resistance-provoking molecules and undersecrete insulin-sensitizing cytokines [30, 32, 33].
There is increasing evidence to suggest that chronic inflammation, insulin resistance, type 2 diabetes and atherosclerosis represent different stages of the same process. Release of TNF-α, IL-8 and IL-10 by adipose tissue from individuals with a BMI of 45 kg/m2 is greater than in subjects with a BMI of 32 kg/m2 [34]. The discovery of high circulating levels of several acute-phase proteins and inflammatory cytokines has led to the view that obese individuals are characterized by a state of low-grade inflammation which links obesity to insulin resistance and the metabolic syndrome [3].
Twenty different adipocytokines have been identified so far and are classified into three groups: 1) cytokines produced mainly in white adipose tissue (e.g. leptin, resistin); 2) cytokines produced exclusively by adipocytes of white adipose tissue (e.g. adiponectin) and 3) cytokines produced primarily in other tissues or organs with simultaneous adipose tissue production (e.g. TNF-α) [35]. Adipocytokines may be even classified according to their putative physiological role into two groups: ‘insulin resistance-inducing factors’ such as resistin, TNF-α, IL-6, PAI-1, angiotensinogen, adipsin, acylation-stimulating protein (ASP) and retinol binding protein-4 (RBP-4) and ‘insulin-sensitizing factors’ such as adiponectin, leptin [35], visfatin [36] and omentin [37] (Table 1).
Table
1.
Adipocytokines and their effects on metabolism |
|
 |
|
Legend:
* Stromal vascular fraction: pre-adipocytes, fibroblasts, endothelial cells, smooth muscle cells, pericytes, monocytes and macrophages. |
|
The metabolic effects of the adipocytokines are probably mediated by a common pathway involving the activation of 5'AMP-activated protein kinase (AMPK) that represents a sensing mechanism of the energy status of the cells. The activation of AMPK increases β-oxidation and decreases fatty acids esterification to tri-glycerides reducing accumulation of lipids in skeletal muscle, liver and other tissues [3].
Adipocytokines in children born small for gestational age
Low birth weight has been associated with increased risk of developing insulin resistance, type 2 diabetes and cardiovascular diseases in adulthood. The thrifty phenotype hypothesis [38] suggests that the fetus adapts to an adverse intrauterine environment by diverting limited nutrients to favor the survival and development of vital organs, such as the brain, at the expense of growth. Important metabolic pathways would be permanently programmed during fetal life, leading to metabolic disturbances in adulthood [31, 39]. There is evidence suggesting that the degree of weight gain and catch-up growth in BMI in postnatal life correlate with insulin resistance [40-43].
Leptin
Leptin, the ob gene product, is primarily produced and secreted by mature adipocytes and binds to its receptors in the hypothalamus. Leptin serves as a major ‘adipostat’ by repressing food intake and promoting energy expenditure. Leptin secretion correlates with adipose tissue mass and nutritional status, and is greater from subcutaneous than visceral adipose tissue [3]. High leptin levels in obese individuals are thought to be a consequence of leptin resistance. Leptin effects, particularly on energy intake and expenditure, are mediated via hypothalamic pathways, but leptin also exerts a direct action on peripheral tissues including muscle and endocrine pancreas [3] (Figure 3).
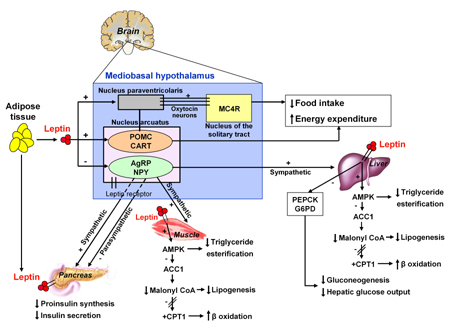 |
|
Figure 3. Central and peripheral actions of leptin. In the hypothalamus, leptin activates neurons located in the arcuate nucleus which express the neuropeptide precursor proopiomelanocortin (POMC) leading to a reduction in food intake and increased energy expenditure [124]. The inhibition of food intake is due to the repression of orexigenic pathways such as neuropeptide Y (NPY) and agouti-related peptide (AgRP), and the activation of anorexigenic pathways such as proopiomelanocortin (POMC) and cocaine and amphetamine-regulated transcript (CART). The food intake-suppressing effects of leptin could be exerted through melanocortin MC4 receptor signaling, by oxytocin neurons which project from the paraventricular nucleus of the hypothalamus into the nucleus of the solitary tract [44, 125]. In muscle, leptin activates 5’AMP-activated protein kinase (AMPK) both directly and indirectly through the activation of hypothalamic-sympathetic axis [126]. AMPK inhibits acetyl CoA carboxilase-1 (ACC1), a key enzyme of the de novo fatty acids synthesis. The inhibition of ACC1 leads to reduced intracellular levels of malonyl CoA [3], the major substrate for long chain fatty acid synthesis that also suppresses fatty acid oxidation by inhibiting carnitine palmitoyltransferase (CPT-1) and acts as a signal molecule in appetite regulation [127-129]. Thereafter, fatty acids are allowed to enter the mitochondria where they undergo β oxidation [3]. Ultimately, leptin promotes fatty acid oxidation and reduces ectopic fat accumulation in non-fat tissues, increasing insulin sensitivity [3, 130]. Finally, the suppressive effect of leptin on insulin production is mediated by both the autonomic nervous system and direct actions via leptin receptor on β-cells [44]. |
|
The existence of an 'adipo-insular axis', describing the insulin promotion of leptin secretion through activation of ob gene transcription has recently been proposed. Leptin, in turn, is able to inhibit insulin release (Figure 4) through central and direct action on β-cell insulin synthesis [44, 45] (Figure 3). Consistent with this model, ablation of leptin receptor from β-cells results in enhanced basal insulin secretion and fasting hypoglycemia [46].
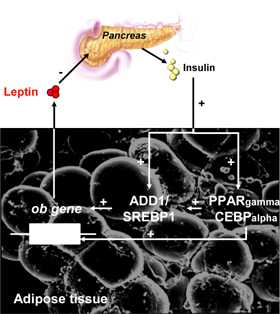 |
|
Figure 4. The adipo-insular axis. The effect of leptin on insulin levels is due to the inhibition of proinsulin synthesis by both central actions and direct actions on β-cells [45]. In turn, insulin plays a crucial role in the regulation of leptin gene expression in white adipose tissue. A candidate transcription factor, adipocyte determination- and differentiation-dependent factor 1/sterol regulatory element binding protein 1 (ADD1/SREBP1), links changes in insulin levels to ob gene expression in mice [131]. ADD1/SREBP1 expression increases after treatment with insulin and transactivates the leptin gene. Conversely, as fat stores increase, rising plasma leptin concentrations would reduce insulin levels, diminishing the formation of adipose tissue. |
|
In SGA children, increased leptin levels have been reported in cord blood [47] and in the first year of life, independently of BMI [48]. High serum leptin values with a loss of the regulatory effect of BMI and gender, suggest that SGA children develop an adaptative leptin resistance beneficial for their catch-up growth [48]. Nonetheless, Martinez-Cordero et al. found lower cord blood leptin levels in SGA than adequate for gestational age (AGA) infants [49]. Furthermore, in the Haguenau study, leptin levels were found to be decreased in young adults born SGA, even after correction for gender, BMI and hyperinsulinemia [31]. This could be due to a greater adiposity in adults born SGA.
Adiponectin
Unlike the other adipocytokines, adiponectin levels correlate inversely with body mass [50, 51]. Recent studies have shown that insulin reduces levels of adiponectin mRNA in a dose- and time-dependent way [52]. Also β-adrenergic agonists [53] and glucocorticoids inhibit adiponectin gene expression and secretion, suggesting that decreased adiponectin production could play a role in catecholamine- and glucocorticoid-induced insulin resistance. In contrast to most adipocytokines, adiponectin expression and serum concentrations are not increased but reduced in a variety of obese and insulin resistant states. In obese rats, a reduction of adiponectin mRNA is present only in the visceral tissue [54]. A reduction in body weight achieved by food restriction is capable of increasing adiponectin mRNA levels in visceral adipose tissue [54]. Human adiponectin gene expression is lower in the visceral than in the subcutaneous adipose tissue of lean subjects [55]. Even in adipocytes differentiated in vitro from precursor mesenchymal cells, adiponectin expression is lower in visceral adipocytes than subcutaneous adipocytes [56].
Several studies have examined human adiponectin gene expression in the adipose tissue of obese subjects, but the results have been inconsistent. Some reports found a reduction in mRNA adiponectin in subcutaneous adipose tissue [57, 58], or in both subcutaneous and visceral adipose tissue depots [55]. Nonetheless, there is an inverse correlation between serum adiponectin and BMI, and individuals with high adiponectin concentrations are at lower risk of developing type 2 diabetes [59].
The adiponectin gene is located on chromosome 3, very close to the diabetes susceptibility locus, and plays a role in the development of type 2 diabetes [60]. Adiponectin levels inversely correlate with a risk of type 2 diabetes. Administration of adiponectin causes glucose-lowering effects and ameliorates insulin resistance in mice [61, 62]. In addition, adiponectin has anti-atherogenic properties, inhibiting monocyte adhesion to endothelial cells and macrophage transformation into foam cells in vitro [63].
We previously reported significantly reduced adiponectin levels in SGA children [60]. Adiponectin was inversely related to height, corrected stature, weight and BMI and directly related to birth weight [60]. In subsequent studies, adiponectin has been reported to be reduced [31, 64, 65], unchanged [49, 66, 67] or increased [67, 68] in SGA subjects.
Resistin
Resistin is a protein expressed in preadipocytes undergoing differentiation [69]. In patients with type 2 diabetes or obesity, both resistin levels and resistin expression in fat cells are increased [66, 70], correlating with hepatic, but not muscle, insulin resistance [71]. In humans, the major source of resistin is the immune cells rather than the adipocytes [68, 72], resistin being a potent inflammatory agent [73]. Insulin inhibits resistin expression in adipocytes [74, 75]. Therefore, the elevated basal plasma resistin levels found in patients with type 2 diabetes [76, 77], despite increased insulin concentrations, may be the result of adipocyte insulin resistance. Resistin inhibits the phosphorylation of hepatic AMPK, decreasing β oxidation and increasing fatty acid esterification in triglycerides, and eventually leading to lipid accumulation [78]. Resistin levels have been reported to be reduced in short SGA children [79].
Tumor necrosis factor-α
TNF-α is implicated in insulin resistance as follows. TNF-α is overexpressed in adipose tissue in obesity and affects insulin signaling by modulating insulin receptor substrate-1 (IRS-1) function via serine phosphorylation [80]. It also affects GLUT4 expression [81], and reduces adiponectin expression in adipose tissue [82]. Finally, it stimulates lipolysis and activates the inflammatory pathways [83]. TNF-α knockout mice do not undergo the development of insulin resistance induced by a high-fat diet, suggesting a role for this cytokine in mediating FFA-induced insulin resistance [84].
In SGA subjects, Jaquet et al. demonstrated an association between higher fasting insulin-to-glucose ratios and polymorphism in TNF-α/-308A [85]. Results for fasting insulin concentration and insulin excursion under OGTT were comparable [85]. These results were subsequently confirmed [86], suggesting that adipose tissue plays a key role in the interaction between intrauterine environment and genetic predisposition in the development of insulin resistance.
In conclusion, data on the adipocytokines in SGA subjects are still contradictory and inconclusive (Table 2).
Table
2.
Adipocytokines in SGA subjects |
|
 |
|
Fat partitioning and lipolysis
Obese patients with an excess of visceral adipose tissue have elevated plasma C-reactive protein concentrations associated with elevated IL-6 and TNF-α levels and reduced adiponectin concentrations [7]. Expanded visceral adipose depot not only leads to altered NEFA metabolism but also to a proinflammatory profile that might contribute to the insulin resistance and altered glucose homeostasis in viscerally obese patients.
In addition, the excess of intra-abdominal fat may represent a marker of the relative inability of subcutaneous adipose tissue to act as an 'energy sink' in a calorie surplus state because of excess energy intake and/or reduced energy expenditure. This would result in ectopic fat deposition in liver, skeletal muscle, heart, β-cells and even visceral adipose tissue [7].
Transgenic fatless mice show liver and muscle insulin resistance and eventually develop diabetes [87]. Surgical implantation of adipose tissue in these mice improves liver and muscle insulin sensitivity [88]. Mice with implantation of subcutaneous adipose tissue in the intra-abdominal cavity show reduced weight, improved insulin sensitivity, reduced body fat percentage, reduced fat mass, reduced adiponectin and leptin [89]. In humans, the severe insulin resistance found in patients with lipodystrophic conditions [90] is also consistent with the role of subcutaneous adipose tissue as a depot buffering the energy excess [91]. Therefore, visceral obesity may also be a marker of defective fat partitioning between adipose tissue, skeletal muscle, liver and heart. There are some differences in the mechanism of accumulation and/or metabolism between visceral and subcutaneous adipose tissue [92].
In db/db mice, four weeks of beta 3-adrenoceptor agonist administration decreased body weight gain and reduced white fat [93]. The Iwao study, using the microdialysis technique in Wistar rats, showed that the lipolysis induced by isoproterenol (a β-adrenergic agonist) is higher in mesenteric than in abdominal subcutaneous adipose tissue at all isoproterenol concentrations, while basal lipolysis was identical in the two depots [94].
Omental fat has been reported to be relatively resistant to insulin suppression of lipolysis in vitro and sensitive to β3-adrenergic stimulation of lipolysis [95]. In vivo studies confirmed that omental adipose tissue is insulin-resistant, even if it extracts one fourth of the insulin passing through. The non-suppressible omental lipolysis in the postprandial state could explain why abdominal obesity is strongly associated with hepatic insulin resistance, since elevated plasma FFA are able to reach the liver [96].
SGA and visceral adiposity
Reduced fetal growth is associated with increased risk of developing insulin resistance, metabolic syndrome or one of its components (hypertension, dyslipidemia, impaired glucose tolerance and type 2 diabetes) and cardiovascular disease in adulthood [97-100].
Insulin resistance has also been detected in young adults and children born SGA [101-105], even in early life [106]. Metabolic syndrome has been found in young adults born SGA: in the Haguenau cohort, at 22 years of age 2.3% of individuals born SGA were affected by the metabolic syndrome compared with only 0.3% in the AGA group [31].
Recent observations emphasize that not only fetal growth but also early postnatal life is critical for the development of insulin resistance and metabolic syndrome. SGA children who experience catch-up growth in childhood are at risk of metabolic consequences. A longitudinal study of a Finnish cohort reported that the highest death rate from coronary heart disease occurred in boys who were thin at birth but whose weight caught up so that they had an average or above average body mass from the age of 7 years [107].
The Hertfordshire study group has recently demonstrated in a case-control study of 32 men of 64-72 years old with a low or high birth weight using dual-energy X-ray absorptiometry (DXA) that, after adjustment for weight and height, the low birth weight group had a higher percentage body fat and fat mass, but lower fat-free soft tissue, muscle mass and muscle-to-fat ratio. Lifelong differences in body composition and fat distribution between the low and high birth weight groups are consistent with programming in early life [40].
Other surveys have shown that catch-up growth in BMI is associated with higher fasting plasma insulin concentration, insulin resistance, and increased total and LDL cholesterol in childhood [41], and correlates with adiposity [43]. SGA children who experienced catch-up growth have a redistribution of fat tissue. Ong and colleagues found that SGA children who showed catch-up growth between zero and two years were fatter and had more central fat distribution at five years than SGA children without catch-up growth [108].
By using DXA, Ibanez et al. showed that SGA children exhibit greater accumulation of total body and abdominal fat than AGA children between the ages of 2 and 4 years, despite having already completed their catch-up growth between birth and 2 years of age [109].
In the Haguenau cohort [31], BMI catch-up growth was significantly related to insulin resistance and metabolic syndrome. However, individuals who experienced greater catch-up were not as obese as young adults, suggesting that fat distribution is more crucial than weight [31]. It has been shown that, after weight loss (chronic diseases, famine, voluntary slimming, fetal or neonatal growth retardation), the subsequent catch-up growth induces excessive fat deposition during nutritional rehabilitation with a disproportionate rate of fat deposition relative to lean tissue recovery. There is an exacerbated suppression of energy expenditure and increased body fat gain during refeeding on a high-fat diet [110-112]. Body fat has a greater energy density and lower energy cost in terms of synthesis/maintenance than proteins. It would provide the organism with a greater capacity to rebuild rapidly an efficient energy reserve, and hence to optimize its survival capacity in the face of recurrent shortage of food [110].
Intrauterine regulation of adipogenesis may be an important mechanism involved in the fetal origins of diabetes and cardiovascular diseases. Studying the differentiation of visceral adipose tissue in SGA children would help to unravel whether a fetal programming of adipogenesis is responsible for the high risk of developing metabolic syndrome in adulthood. Adipocytic mesenchymal stem cells retain the capacity to differentiate into fat cells that maintain human-specific adipocyte characteristics (Figure 5).
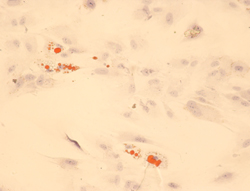 |
|
Figure 5. Adipocytic differentiation of adipose-derived stem cells. Cells grown in adipogenic medium after two weeks assume a rounded shape and lipidic droplets in the cytosol that stained positively with Oil-Red-O, lipid dye. |
|
Adipose-derived stem cells (ADSC) could be more prone in SGA subjects to differentiate into a functionally different visceral adipose tissue after birth. The study of the differentiation capacity of visceral fat could help to understand the mechanisms involved in the pathogenesis of metabolic syndrome. Interventions aimed at normalizing fat partitioning in childhood could prevent insulin resistance and metabolic syndrome in subjects born small for gestational age.
References
- Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell 2001. 104:531-543. [DOD] [CrossRef]
- Saltiel AR. You are what you secrete. Nat Med 2001. 7:887-888. [DOD] [CrossRef]
- Vettor R, Milan G, Rossato M, Federspil G. Review article: adipocytokines and insulin resistance. Aliment Pharmacol Ther 2005. 22(Suppl 2):3-10. [DOD] [CrossRef]
- Harris MI, Flegal KM, Cowie CC, Eberhardt MS, Goldstein DE, Little RR, Wiedmeyer HM, Byrd-Holt DD. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The third National Health and Nutrition Examination Survey 1988-1994. Diabetes Care 1998. 21:518-524. [DOD] [CrossRef]
- Pi-Sunyer F. Weight and non-insulin-dependent diabetes mellitus. Am J Clin Nutr 1996. 63(Suppl 3):426S-429S. [DOD]
- Knowler WC, Pettitt DJ, Savage PJ, Bennett PH. Diabetes incidence in Pima Indians: contributions of obesity and parental diabetes. Am J Epidemiol 1981. 113:144-156. [DOD]
- Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature 2006. 444:881-887. [DOD] [CrossRef]
- DeFronzo RA. Lilly Lecture 1987. The triumvirate: cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988. 37:667-687. [DOD]
- Chan JM, Rimm EB, Colditz GA, Stampfer MJ, Willett WC. Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care 1994. 14:961-969. [DOD] [CrossRef]
- Bjorntorp P. Metabolic implications of body fat distribution. Diabetes care 1991. 14:1132-1143. [DOD] [CrossRef]
- Kissebah AH, Peiris AN. Biology of regional body fat distribution: relationship to non-insulin-dependent diabetes mellitus. Diabetes Metab Rev 1989. 5(2):83-109. [DOD]
- Arner P. Regional adipocity in man. J Endocrinol 1997. 155:191-192. [DOD] [CrossRef]
- Zierath JR, Livingston JN, Thörne A, Bolinder J, Reynisdottir S, Lönnqvist F, Arner P. Regional difference in insulin inhibition of non-esterified fatty acid release from human adipocytes: relation to insulin receptor phosphorylation and intracellular signaling through the insulin receptor substrate-1-pathway. Diabetologia 1998. 41(11):1343-1354. [DOD] [CrossRef]
- Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 hours in patients with NIDDM. Diabetes 1988. 37:1020-1024. [DOD] [CrossRef]
- Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes, defining their role in the development of insulin resistance and cell dysfunction. Eur J Clin Invest 2002. 32(Suppl 3):14-23. [DOD] [CrossRef]
- Golay A, Felber JP, Jequier E, DeFronzo RA, Ferrannini E. Metabolic basis of obesity and noninsulin-dependent diabetes mellitus. Diabetes Metab Rev 1988. 4:727-747. [DOD]
- Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997. 46:3-10. [DOD] [CrossRef]
- McGarry JD. Bantig Lecture 2001. Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002. 51:7-18. [DOD] [CrossRef]
- Eldor R, Raz I. Lipotoxicity versus adipotoxicity - the deleterious effects of adipose tissue on beta cells in the pathogenesis of type 2 diabetes. Diabetes Res Clin Pract 2006. 74(2 Suppl 1):S3-S8. [DOD] [CrossRef]
- Goodpaster BH, Thaete FL, Kelley BE. Thigh adipose tissue distribution is associated with insulin resistance in obesity and type 2 diabetes mellitus. Am J Clin Nutr 2000. 71:885-892. [DOD]
- Greco AV, Mingrone G, Giancaterini A, Manco M, Morroni M, Cinti S, Granzotto M, Vettor R, Camastra S, Ferrannini E. Insulin resistance in morbid obesity. Reversal with intramyocellular fat depletion. Diabetes 2002. 51:144-151. [DOD] [CrossRef]
- Seppälä-Lindroos A, Vehkavaara S, Häkkinen AM, Goto T, Westerbacka J, Sovijärvi A, Halavaara J, Yki-Järvinen H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 2002. 87(7):3023-3028. [DOD] [CrossRef]
- Simha V, Garg A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol 2006. 17(2):162-169. [DOD] [CrossRef]
- DeFronzo RA. Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int J Clin Pract 2004. 58(Suppl 143):9-21. [DOD] [CrossRef]
- DeFronzo RA. Pathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetes genes. Diabetes Rev 1997. 3:177-269. [DOD]
- Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM: genetic and clinical implications. Diabetes 1995. 44:863-870. [DOD] [CrossRef]
- DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in non-insulin dependent (type II) diabetes mellitus. J Clin Invest 1985. 76(1):149-155. [DOD]
- Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, DeFronzo RA. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest 1989. 84:205-213. [DOD]
- DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non-insulin dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism 1989. 38:387-395. [DOD] [CrossRef]
- Sewter CP, Blows F, Vidal-Puig A, O'Rahilly S. Regional differences in the response of human pre-adipocytes to PPARgamma and RXRalpha agonists. Diabetes 2002. 51(3):718-723. [DOD] [CrossRef]
- Levy-Marchal C, Czernichow P. Small for gestational age and the metabolic syndrome: which mechanism is suggested by epidemiological and clinical studies? Horm Res 2006. 65(Suppl 3):123-130. [DOD]
- Fruhbeck G, Gomez-Ambrosi J, Muruzabal FJ, Burrell MA. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am J Physiol 2001. 280:E827-E847. [DOD]
- Motoshima H, Wu X, Sinha MK, Hardy VE, Rosato EL, Barbot DJ, Rosato FE, Goldstein BJ. Differential regulation of adiponectin secretion from cultured human omental and subcutaneous adipocytes: effects of insulin and rosiglitazone. J Clin Edocrinol Metab 2002. 87:5662-5667. [DOD] [CrossRef]
- Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, Adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004. 145(5):2273-2282. [DOD] [CrossRef]
- Housa D, Housova J, Vernerova Z, Haluzik M. Adipocytokines and cancer. Physiological Research 2005. 55(3):233-244. [DOD]
- Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, et al. Visfatin: a protein secreted by visceral fat that mimics the effect of insulin. Science 2005. 307(5708):426-430. [DOD] [CrossRef]
- Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, Shuldiner AR, Fried SK, McLenithan JC, Gong DW. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab 2006. 290:E1253-E1261. [DOD] [CrossRef]
- Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 1992. 35:595-601. [DOD] [CrossRef]
- Saenger P, Czernichow P, Hughes I, Reiter EO. Small for gestational age: short stature and beyond. Endocr Rev 2007. 28(2):219-251. [DOD] [CrossRef]
- Kensara OA, Wootton SA, Phillips DI, Patel M, Jackson AA, Elia M, Hertfordshire Study Group. Fetal programming of body composition: relation between birth weight and body composition measured with dual-energy X-ray absorptiometry and anthropometric methods in older Englishmen. Am J Clin Nutr 2005. 85(5):980-987. [DOD]
- Yajnik CS. Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. J Nutr 2004. 134:205-210. [DOD]
- Ezzahir N, Alberti C, Deghmoun S, Zaccaria I, Czernichow P, Levy-Marchal C, Jaquet D. Time course of catch-up in adiposity influences adult anthropometry in individuals who were born small for gestational age. Pediatr Res 2005. 58:243-247. [DOD] [CrossRef]
- Hediger ML, Overpeck MD, Kuczmarski RJ, McGlynn A, Maurer KR, Davis WW. Muscularity and fatness of infants and young children born small- or large-for-gestational-age. Pediatrics 1998. 102(5):E60. [DOD] [CrossRef]
- Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006. 444:847-853. [DOD] [CrossRef]
- Kieffer TJ, Habener JF. The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am J Physiol Endocrinol Metab 2000. 278(1):E1-E14. [DOD]
- Covey SD, Wideman RD, McDonald C, Unniappan S, Huynh F, Asadi A, Speck M, Webber T, Chua SC, Kieffer TJ. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab 2006. 4(4):291-302. [DOD] [CrossRef]
- Shekhawat PS, Garland JS, Shivpuri C, Mick GJ, Sasidharan P, Pelz CJ, McCormick KL. Neonatal cord blood leptin: its relationship to birth weight, body mass index, maternal diabetes, and steroids. Pediatr Res 1998. 43:338-343. [DOD] [CrossRef]
- Jaquet D, Leger J, Tabone MD, Czernichow P, Levy-Marchal C. High serum leptin concentrations during catch-up growth of children born with intrauterine growth retardation. J Clin Endocrinol Metab 1999. 84:1949-1953. [DOD] [CrossRef]
- Martinez-Cordero C, Amador-Licona N, Guizar-Mendoza JM, Hernandez-Mendez J, Ruelas-Orozco G. Body fat at birth and cord blood levels of insulin, adiponectin, leptin, and insulin-like growth factor-1 in small-for-gestational-age infants. Arch Med Res 2006. 37:490-494. [DOD] [CrossRef]
- Yatagai T, Nagasaka S, Taniguchi A, Fukushima M, Nakamura T, Kuroe A, Nakai Y, Ishibashi S. Hypoadiponectinemia is associated with visceral fat accumulation and insulin resistance in Japanese men with type 2 diabetes mellitus. Metabolism 2003. 52:1274-1278. [DOD] [CrossRef]
- Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, Matsuzawa Y. Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes 2001. 50:1126-1133. [DOD] [CrossRef]
- Fasshauer M, Klein J, Lossner U, Paschke R. Negative regulation of adipose-expressed galectin-12 by isoproterenol, tumor necrosis factor alpha, insulin and dexamethasone. Eur J Endocrinol 2002. 147:553-559. [DOD] [CrossRef]
- Zhang Y, Matheny M, Zolotukhin S, Tumer N, Scarpace PJ. Regulation of adiponectin and leptin gene expression in white and brown adipose tissues: influence of beta3-adrenergic agonists, retinoic acid, leptin and fasting. Biochim Biophys Acta 2002. 1584(2-3):115-122. [DOD]
- Milan G, Granzotto M, Scarda A, Calcagno A, Pagano C, Federspil G, Vettor R. Resistin and adiponectin expression in visceral fat of obese rats: effect of weight loss. Obes Res 2002. 10:1095-1103. [DOD]
- Lihn AS, Bruun JM, He G, Pedersen SB, Jensen PF, Richelsen B. Lower expression of adiponectin mRNA in visceral adipose tissue in lean and obese subjects. Mol Cell Endocrinol 2004. 219(1-2):9-15. [DOD] [CrossRef]
- Perrini S, Laviola L, Cignarelli A, Melchiorre M, De Stefano F, Caccioppoli C, Natalicchio A, Orlando MR, Garruti G, De Fazio M, et al. Fat depot-related differences in gene expression, adiponectin secretion, and insulin action and signalling in human adipocytes differentiated in vitro from precursor stromal cells. Diabetologia 2007. In press. [DOD]
- Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 2003. 52:1779-1785. [DOD] [CrossRef]
- Degawa-Yamauchi M, Moss KA, Bovenkerk JE, Shankar SS, Morrison CL, Lelliott CJ, Vidal-Puig A, Jones R, Considine RV. Regulation of adiponectin expression in human adipocytes: effects of adiposity, glucocorticoids, and tumor necrosis factor. Obes Res 2005. 13(4):662-669. [DOD]
- Spranger J, Kroke A, Möhlig M, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF. Adiponectin and protection against type 2 diabetes mellitus. Lancet 2003. 361(9353):226-228. [DOD] [CrossRef]
- Cianfarani S, Martinez C, Maiorana A, Scire G, Spadoni GL, Boemi S. Adiponectin levels are reduced in children born small for gestational age and are inversely related to postnatal catch-up growth. J Clin Endocrinol Metab 2004. 89:1346-1351. [DOD] [CrossRef]
- Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 2001. 7(8):947-953. [DOD] [CrossRef]
- Yamauchi T, Kamon J, Waki J, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-kasaoka N, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 2001. 7:941-946. [DOD] [CrossRef]
- Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y, Ishigami M, Kuriyama H, Kishida K, Nishizawa H, et al. Adipocyte-derived plasma protein, adiponectin, suppressed lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 2001. 103:1057-1063. [DOD]
- Iniguez G, Soto N, Avila A, Salazar T, Ong K, Dunger D, Mericq V. Adiponectin levels in the first two years of life in a prospective cohort: relations with weight gain, leptin levels and insulin sensitivity. J Clin Endocrinol Metab 2004. 89(11):5500-5503. [DOD] [CrossRef]
- Klamer A, Skogstrand K, Hougaard DM, Norgaard-Pedersen B, Juul A, Greisen G. Adiponectin levels measured in dried blood spot from neonates born small and appropriate for gestational age. Eur J Endocrinol 2007. 157:189-194. [DOD] [CrossRef]
- Ibanez L, Fucci A, Valls C, Ong K, Dunger D, de Zegher F. Neutrophil count in small for gestational age children: contrasting effects of metformin and growth hormone therapy. J Clin Endocrinol Metab 2005. 90:3435-3439. [DOD] [CrossRef]
- Evagelidou EN, Giapros VI, Challa AS, Kiortsis DN, Tsatsoulis AA, Andronikou SK. Serum adiponectin levels, insulin resistance, and lipid profile in children born small for gestational age are affected by the severity of growth retardation at birth. Eur J Endocrinol 2007. 156(2):271-277. [DOD] [CrossRef]
- Lopez-Bermejo A, Casano-Sancho P, Fernandez-Real JM, Kihara S, Funahashi T, Rodriguez-Hierro F, Ricart W, Ibanez L. Both intrauterine growth restriction and postnatal growth influence childhood serum concentrations of adiponectin. Clin Endocrinol (Oxf) 2004. 61:339-346. [DOD] [CrossRef]
- Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature 2001. 409:307-312. [DOD] [CrossRef]
- Wang H, Chu WS, Hemphill C, Elbein SC. Human resistin gene: molecular scanning and evaluation of association with insulin sensitivity and type 2 diabetes in Caucasians. J Clin Endocrinol Metab 2002. 87(6):2520-2524. [DOD] [CrossRef]
- Rajala MW, Obici S, Scherer PE, Rossetti L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest 2003. 111:225-230. [DOD] [CrossRef]
- Savage DB, Sewter CP, Klenk ES, Segal DG, Vidal-Puig A, Considine RV, O'Rahilly S. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes 2001. 50(10):2199-2202. [DOD] [CrossRef]
- Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 1995. 270:26746-26749. [DOD] [CrossRef]
- Hartman HB, Hu X, Tyler KX, Dalal CK, Lazar MA. Mechanisms regulating adipocyte expression in of resistin. J Biol Chem 2002. 277:19754-19761. [DOD] [CrossRef]
- Haugen F, Jorgensen A, Drevon CA, Trayhurn P. Inibition by insulin of resistin gene expression in 3T3-L1 and mouse adipose cells. FEBS Lett 2001. 507:105-108. [DOD] [CrossRef]
- McTernan CL, McTernan PG, Harte AL, Levick PL, Barnett AH, Kumar S. Resistin, central obesity, and type 2 diabetes. Lancet 2002. 359(9300):46-47. [DOD] [CrossRef]
- Bajaj M, Suraamornkul S, Hardies LJ, Pratipanawatr T, DeFronzo RA. Plasma resistin concentration, hepatic fat content, and hepatic and peripheral insulin resistance in pioglitazone-treated type 2 diabetic patients. Intern J Obes Relat Metab Disord 2004. 28:783-789. [DOD] [CrossRef]
- Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, et al. Regulation of fasted blood glucose by resistin. Science 2004. 303(5661):1195-1198. [DOD] [CrossRef]
- Willemsen RH, van Dijk M, de Rijke YB, van Toorenenbergen AW, Mulder PG, Hokken-Koelega AC. Effect of growth hormone therapy on serum adiponectin and resistin levels in short, small-for-gestational-age children and associations with cardiovascular risk parameters. J Clin Endocrinol Metab 2007. 92:117-123. [DOD] [CrossRef]
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem 1993. 268(35):26055-26058. [DOD]
- Stephens JM, Lee J, Pilch PF. Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem 1997. 272:971-976. [DOD] [CrossRef]
- Ruan H, Miles PD, Ladd CM, Ross K, Golub TR, Olefsky JM, Lodish HF. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes 2002. 51:3176-3188. [DOD] [CrossRef]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993. 259:87-91. [DOD] [CrossRef]
- Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997. 389:610-614. [DOD] [CrossRef]
- Jaquet D, Tregouet DA, Godefroy T, Nicaud V, Chevenne D, Tiret L, Czernichow P, Levy-Marchal C. Combined effects of genetic and environmental factors on insulin resistance associated with reduced fetal growth. Diabetes 2002. 51(12):3473-3478. [DOD] [CrossRef]
- Casano-Sancho P, Lopez-Bermejo A, Fernandez-Real JM, Monros E, Valls C, Rodriguez-Gonzalez FX, Ricart W, Ibanez L. The tumour necrosis factor (TNF)-alpha-308GA promoter polymorphism is related to prenatal growth and postnatal insulin resistance. Clin Endocrinol (Oxf) 2006. 64(2):129-135. [DOD] [CrossRef]
- Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 2000. 275:8456-8460. [DOD] [CrossRef]
- Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, Vinson C, Eckhaus M, Reitman ML. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest 2000. 105:271-278. [DOD]
- Kahn R. Personal communication. NIH conference, January 2007. [DOD]
- Garg A, Misra A. Lipodystrophies: rare disorders causing metabolic syndrome. Endocr Metab Clin North Am 2004. 33:305-331. [DOD] [CrossRef]
- Danforth E Jr. Failure of adipocytic differentiation causes type II diabetes mellitus? Nat Genet 2000. 26(1):13. [DOD]
- Rebuffe-Scrive M, Andersson B, Olbe L, Bjorntorp P. Metabolism of adipose tissue in intraabdominal depots of nonobese men and women. Metabolism 1989. 38:453-458. [DOD] [CrossRef]
- Hashimoto K, Nagao Y, Ida K, Takeda M, Murakami N, Kato K, Mizota M. Improvement of metabolic disorders and visceral fat obesity by the beta-3-adrenoreceptor agonist (R*,R*)-(+/-)-methyl-4-[2-[2-hydroxy-2-(3-chlorophenyl)ethylamino]propyl]-phenoxyacetate hydrobromide (BRL35135A) in genetically obese rodents. Biochem Pharmacol 1996. 52(10):1529-1535. [DOD] [CrossRef]
- Iwao N, Oshida Y, Sato Y. Regional difference in lipolysis caused by a beta-adrenergic agonist as determined by the microdialysis technique. Acta Physiol Scand 1997. 161:481-487. [DOD] [CrossRef]
- Zierath JR, Livingston JN, Thorne A, Bolinder J, Reynisdottir S, Lonnqvist F, Arner P. Regional difference I insulin inhibition of non-esterified fatty acid release from human adipocytes: relation to insulin receptor phosphorylation and intracellular signalling through the insulin receptor substrate-1 pathway. Diabetologia 1998. 41:1343-1354. [DOD] [CrossRef]
- Mittelman SD, Van Citters GW, Kirkman EL, Bergman RN. Extreme insulin resistance of the central adipose depot in vivo. Diabetes 2002. 51:755-761. [DOD] [CrossRef]
- Barker DJ, Winder PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 1989. 2:577-580. [DOD] [CrossRef]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. Br Med J 1991. 303:1019-1022. [DOD]
- Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 1993. 36(1):62-67. [DOD] [CrossRef]
- Barker DJ. The intrauterine origins of cardiovascular disease. Acta Paediatr Suppl 1993. 82(Suppl 391):93-99. [DOD]
- Phillips DI, Barker DJ, Hales CN, Hirst S, Osmond C. Thinness at birth and insulin resistance in adult life. Diabetologia 1994. 37:150-154. [DOD] [CrossRef]
- Jaquet D, Gaboriau A, Czernichow P, Levy-Marchal C. Insulin resistance early in adulthood in subjects born with intrauterine growth retardation. J Clin Endocrinol Metab 2000. 85:1401-1406. [DOD] [CrossRef]
- Flanagan DE, Moore VM, Godsland IF, Cockington RA, Robinson JS, Phillips DI. Fetal growth and the physiological control of glucose tolerance in adults: a minimal model analysis. Am J Physiol Endocr Metab 2000. 85:1401-1406. [DOD]
- Hofman PL, Cutfield WS, Robinson EM, Bergman RN, Menon RK, Sperling MA, Gluckman PD. Insulin resistance in short children with intrauterine growth retardation. J Clin Endocrinol Metab 1997. 82:402-406. [DOD] [CrossRef]
- Veening MA, Van Weissenbruch MM, Delemarre-Van De Waal HA. Glucose tolerance, insulin sensitivity, and insulin secretion in children born small for gestational age. J Clin Endocrinol Metab 2002. 87:4657-4661. [DOD] [CrossRef]
- Soto N, Bazaes RA, Pena V, Salazar T, Avila A, Iniguez G, Ong KK, Dunger DB, Mericq MV. Insulin sensitivity and secretion are related to catch-up growth in small-for-gestational-age infants at age 1 year: results from a prospective cohort. J Clin Endocrinol Metab 2003. 88:3645-3650. [DOD] [CrossRef]
- Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. Br Med J 1999. 318:427-431. [DOD]
- Ong KK, Ahmed ML, Emmett PM, Preece MA, Dunger DB, Avon Longitudinal Study of Pregnancy and Childhood Study Team. Association between postnatal catchup growth and obesity in childhood: prospective cohort study. Br Med J 2000. 320:967-971. [DOD] [CrossRef]
- Ibanez L, Ong K, Dunger DB, de Zegher F. Early development of adiposity and insulin resistance after catch-up weight gain in small-for-gestational-age children. J Clin Endocrinol Metab 2006. 91(6):2153-2158. [DOD] [CrossRef]
- Dulloo AG, Jacquet J, Montani JP. Pathways from weight fluctuations to metabolic diseases: focus on maladaptive thermogenesis during catch-up fat. Int J Obes 2002. 26(Suppl 2):S46-S57. [DOD] [CrossRef]
- Dulloo AG, Jacquet J, Seydoux J, Montani JP. The thrifty 'catch-up fat' phenotype: its impact on insulin sensitivity during growth trajectories to obesity and metabolic syndrome. Int J Obes (Lond) 2006. 30(Suppl 4):S23-S35. [DOD] [CrossRef]
- Dulloo AG. Regulation of fat storage via suppressed thermogenesis: a thrifty phenotype that predisposes individuals with catch-up growth to insulin resistance and obesity. Horm Res 2006. 65(Suppl 3):90-97. [DOD] [CrossRef]
- Exton JH, Corbin JG, Park CR. Control of gluconeogenesis in liver. IV. Differential effects of fatty acids and glucagons on ketogenesis and gluconeonesis in the perfused rat liver. J Biol Chem 1969. 244(15):4095-4102. [DOD]
- Massillon D, Barzilai N, Hawkins M, Prus-Wertheimer D, Rossetti L. Induction of hepatic glucose-6-phosphatase gene expression by lipid infusion. Diabetes 1997. 46:153-157. [DOD] [CrossRef]
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963. 1:785-789. [DOD] [CrossRef]
- De Fea K, Roth RA. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry 1997. 36:12939-12947. [DOD] [CrossRef]
- Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PBK pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 1999. 274:24202-24210. [DOD] [CrossRef]
- Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid induced cell apoptosis. Proc Natl Acad Sci USA 1998. 95:2498-24502. [DOD] [CrossRef]
- McGarry JD, Dobbins RL. Fatty acids, lipotoxicity and insulin secretion. Diabetologia 1999. 42:128-138. [DOD] [CrossRef]
- Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest 1994. 93:870-876. [DOD]
- Zhou YP, Grill V. Long term exposure to fatty acids and ketones inhibits B-cell functions in human pancreatic islets of Langerhans. J Clin Endocrinol Metab 1995. 80(5):1584-1590. [DOD] [CrossRef]
- Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patane G, Boggi U, Piro S, Anello M, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets. Evidence that cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002. 51:1437-1442. [DOD] [CrossRef]
- Mason TM, Goh T, Tchipashvili V, Sandhu H, Gupta N, Lewis GF, Giacca A. Prolonged elevation of plasma free fatty acids desensitizes the insulin secretory response to glucose in vivo in rats. Diabetes 1999. 48:524-530. [DOD] [CrossRef]
- Broberger C. Brain regulation of food intake and appetite: molecules and network. J Int Med 2005. 258:301-327. [DOD] [CrossRef]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005. 123:493-505. [DOD] [CrossRef]
- Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002. 415:339-343. [DOD] [CrossRef]
- Hu Z, Cha SH, Chohnan S, Lane MD. Hypothalamic malonyl-CoA as a mediator of feeding behaviour. Proc Natl Acad Sci USA 2003. 100:12624-12629. [DOD] [CrossRef]
- Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 2004. 3:340-351. [DOD] [CrossRef]
- Ruderman NB, Saha AK, Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology 2003. 144:5166-5171. [DOD] [CrossRef]
- Muoio DM, Dohm GL, Fiedorek FT Jr, Tapscott EB, Coleman RA. Leptin directly alters lipid partitioning in skeletal muscle. Diabetes 1997. 46:1360-1363. [DOD] [CrossRef]
- Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, Lowell BB, Spiegelman BM. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 1998. 101:1-9. [DOD]
This article has been cited by other articles:
|


)
)
)
)
)
)
)






